×
![]()
Corresponding Author: Richard Kolesnick
Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10065 (USA)
Tel. +1 (646) 888-2174, Fax +1 (646) 422-0281, E-Mail r-kolesnick@ski.mskcc.org
Acid Sphingomyelinase-Ceramide Induced Vascular Injury Determines Colorectal Cancer Stem Cell Fate
Christy Lia Stefan Klinglera Sahra Bodoa Jin Chenga Yan Pana Mohammed Adileha Maria Laura Martina,b John Fullera Regina Feldmana Adam Michelc Zhigang Zhangd Zvi Fukse Richard Kolesnicka
aLaboratory of Signal Transduction Memorial Sloan-Kettering Cancer Center, New York, NY, USA, bEnglander Institute for Precision Medicine, Weill Cornell Medicine, New York, USA, cLaboratory of Comparative Pathology, Rockefeller University, Weill Cornell Medicine and Memorial Sloan-Kettering Cancer Center, New York, NY, USA, dDepartment of Epidemiology and Biostatistics Memorial Sloan-Kettering Cancer Center, New York, NY, USA, eDepartment of Radiation Oncology Memorial Sloan-Kettering Cancer Center, New York, NY, USA
Introduction
Our recent studies into the radiation response of validated normal and tumor tissue stem cell populations indicate preferential use of homologous recombination repair (HRR) to resolve potentially-lethal DNA double stand breaks (DSBs) [1-3]. In C. elegans, germline stem cells repair radiation-induced DSBs exclusively by HRR despite abundant cellular availability of the enzymes of the non-homologous end joining (NHEJ) pathway [1]. Based on this observation, it was suggested that activity of error-prone NHEJ enzymes was being actively suppressed in the C. elegans germline in order to maintain genomic integrity [1-3]. Further, a temperature-sensitive NOTCH mutation that drives formation of a tumor comprised almost entirely of C. elegans germline stem cells, engaged HRR exclusively in response to ionizing radiation. A similar phenotype was observed in human T-cell lymphoblastic lymphoma CUTLL-1 cells grown as a flank chloroma in nude mice [1-3]. As in C. elegans, the well-established Lgr5+ stem cell population in the murine GI tract preferentially uses HRR to repair radiation-induced DSBs compared with its differentiated progeny [4]. Accumulating evidence indicates that radiosensitivity of the Lgr5+ stem cell population in both the large and small intestines is determinant in survival of these organs with the Lgr5+ population of the large intestine being significantly more resistant to radiation lethality than the Lgr5+ population of the radiosensitive small intestine [2-4].
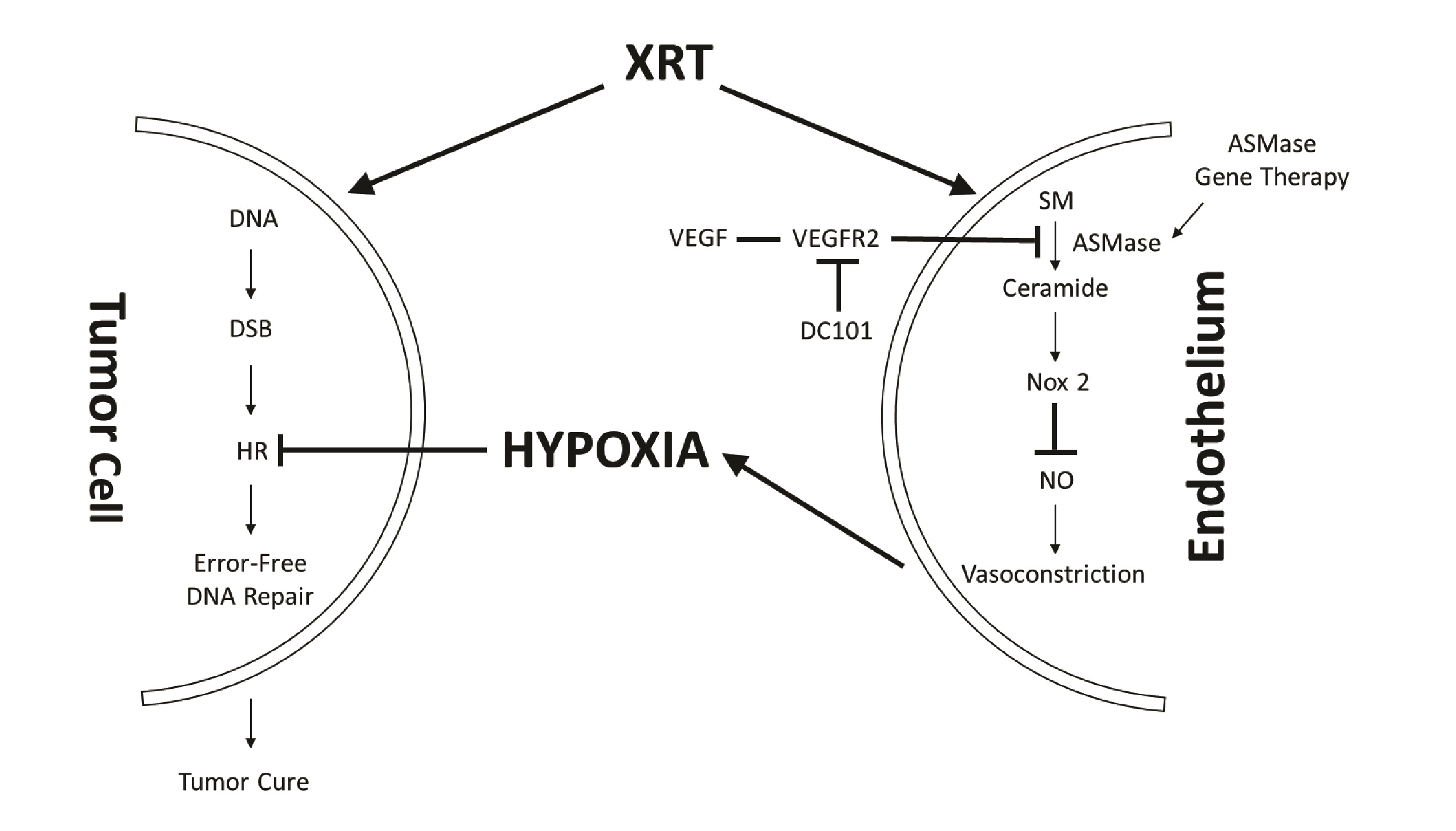
Our program has pioneered the use of a new form of radiation treatment of extracranial tumors, termed single dose radiotherapy (SDRT), which is significantly more effective in human tumor cure than conventional fractionated radiotherapy (95% vs. 65%, respectively) [5-9]. We recently published that this high success of SDRT results from engagement of a new biology that targets HRR repair [10]. In this paradigm (Fig. 1), SDRT, at a threshold of 12Gy, rapidly induces a dose-dependent load of DSBs in parenchymal tissue cells, concomitant with injury to the exposed tumor neo-angiogenic microvascular endothelium. Exposed endothelial cells respond, within seconds, by activating acid sphingomyelinase (ASMase), which hydrolyzes sphingomyelin on the external leaflet of the plasma membrane to generate ceramide, which coalesces, due to biophysical forces (hydrogen bonding, hydrophobic & van der Waal forces), to form a structure we term a ceramide-rich platform (CRP) [11, 12]. CRPs are signaling macrodomains that enable, amongst other functions, a rapid oligomerization in endothelial plasma membranes of the NADPH oxidase-2 (NOX-2) complex [13]. NOX-2 generates superoxide radical redox signaling that depletes the principal microvascular vasodilator nitric oxide (NO) via uncoupling dimeric NO synthase [13, 14], yielding a state of unopposed endothelin vasoconstriction [15]. This acute vasoconstriction results in marked perfusion defects in mouse xenografts and in human spinal metastases, as detected by magnetic resonance imaging [10]. Ensuing tumor hypoxia, widespread throughout the tumor interstitial space, renders oxidative proteotoxic stress within parenchymal tumor clonogens, ultimately activating an adaptive SUMO Stress Response (SSR) that consumes cellular reservoirs of SUMO, including that of SUMO3 at chromatin, which is required for positioning and activation of multiple enzymes of the HRR apparatus [16, 17]. Consequent global HRR inactivation occurring in tumors of all types accounts for the high clinical success of SDRT in local cure of human cancer.
Whereas colorectal cancer (CRC) has as much as a log increase in Lgr5+ stem cell-like cells [18], and whereas normal and tumor stem cells preferentially use HRR to repair radiation-induced DSBs, we developed a radiosensitive and radioresistant patient-derived CRC xenograft model in mice to address whether the high success rate of SDRT might represent specific targeting of the tumor stem cell compartment.
Materials and Methods
Xenograft Transplantation
CLR1-1 and CLR27-2 human CRC PDXs, obtained from the MSKCC Antitumor Assessment Core Facility, were propagated in NOD/scid (ICR- Prkdc/scid; NSG) female mice (Taconic Stock# ICRSC). For experiments, PDXs were cut into small fragments, individual cells and small clusters isolated using a molecular sieve were resuspended in phosphate buffered saline and combined 1:1 (v/v) with Matrigel (BD Biosciences) on ice. Two hundred µg of the tumor cell-Matrigel mixture was injected subcutaneously into the right flank of NSG mice.
Single-Dose Radiotherapy
Animals harboring 100-150 mm3 PDXs were sedated with ketamine, and immobilized in specialized lead jigs that exposed only tumor and surrounding skin to the radiation source. Single dose radiotherapy (SDRT) was administered via a Philips MG-320 X-ray unit at 118.3 cGy/min.
Tumor Response
Tumor volume, based on caliper measurements, was calculated according to the formula of Kim et al. [19]. Tumor cure is defined as no detectable tumor confirmed at autopsy at 90-120 days, while complete response is defined as tumor mass becoming unmeasurable.
DC101 formulation and administration
Rat anti-mouse VEGF receptor 2 IgG1 DC101 (Cat. # BE0060, BioCell) was provided at 8.56mg/ml in phospho-buffered saline. For these studies, animals were treated intravenously with DC101 (1.6mg/25 gm mouse) at 1h prior to irradiation.
ASMase Gene Therapy
When CLR27-2 tumor xenograft host animals reached tumor sizes of 100-150 mm3, 1x1010 PFU of H2E-PPE1(3x)-ASMase construct, which expresses human ASMase exclusively in tumor neo-angiogenic microvasculature, was administered via tail-vein injection, as published by us [20]. SDRT was administered to mice five days thereafter at the time of maximal ASMase expression.
In-Situ Hybridization (ISH) of LGR5 Expression
Paraffin-embedded tissue sections (5µm width) were evaluated using the Affymetrix QuantiGene ViewRNA ISH 1-Plex Assay Kit or ACD Biosciences RNAscope 2.5 HD Assay according to manufacturer’s protocols. Tumor sections stained with Lgr5 ISH were scanned using the Panoramic Flash 250 scanner (Perkin Elmer) and analyzed using Panoramic Viewer software. Images were taken at either 40x or 20x magnification. Lgr5+ cells per field were manually counted in ImageJ, with nuclei containing three or more foci qualifying as “positive”.
TUNEL-MECA-32 Double Staining
Tumor specimens were obtained at the indicated times post SDRT, fixed in 10% buffered formalin phosphate, embedded in paraffin, and 5µm sections were evaluated for endothelial cell apoptosis by double staining with MECA 32 and terminal deoxytransferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL), as published [21, 22].
Tumor Histology
Tumors were stained with H&E and analyzed for presence of residual tumor with the assistance of A.S., a trained mouse pathologist. Residual tumor was scanned using the Panoramic Flash 250 scanner, analyzed with Panoramic Viewer software, and tumor area quantified using QuPath whole slide image analysis software.
Organoid isolation from PDX tumors
After euthanizing PDX-bearing mice, tumors were extracted from the flank area and all connective tissue and blood vessels were cleaned and washed vigorously. Tumor fragments were sliced into 1-3 mm pieces and then suspended into 10 ml of DMEM high glucose containing 1% FBS, 500 U/mL collagenase IV. The mixture was incubated for 30 min at 37 °C in a shaking water bath. Thereafter, tissue fragments were vigorously shaken using a 10 ml pipette for further mechanical disruption, allowed to settle under gravity for 1 min, and the supernatant was removed for inspection by inverted microscopy. The resuspension/sedimentation procedure was repeated three times, liberated tumor tissues in suspension were combined, passed through 100 µm cell strainer to remove muscle material, and then centrifuged at 4 °C at 300 x g for 5 min. Isolated tumor clusters were pelleted, washed with cold 1% FBS/DMEM, and centrifuged three times at 60 x g for 3 min to separate tumor clusters from dead single cells.
Tumor organoid culture
Isolated tumor clusters were resuspended in Matrigel and plated as droplets, covered with advanced DMEM/F12 media containing 1 mmol/L HEPES, 1 mmol/L glutamax, and 100 U/mL antibiotics (ADF) supplemented with B27, N2, and 1 mmol/L N-acetylcysteine (NAC), 50 ng/mL human recombinant EGF, 100 ng/mL mouse recombinant Noggin together with gastrin, nicotinamide, A83-01 and SB202190.
Organoid radiation experiments
Pelleted tumor organoids were dissociated in 2-3 ml of TryplE containing 200 U/mL DNAase, 0.5 mmol/L N-acetylcysteine (NAC), and 10 mmol/L Y-27632 for 3 min at 37 °C in a water bath, shaking every minute to generate a single-cell suspension. Dissociated cells were washed with 1% FBS/DMEM. Cells were counted and plated to form 100-150 tumor organoids per well. Cells were left to mature to a well-formed organoid (3 days after plating). At least three wells were used for each radiation dose. Organoids were exposed to single-fraction radiation (range, 0-10 Gy). Manual counting under an inverted brightfield microscope of surviving organoids at day 7 post-radiation was used to generate classic radiation dose-response curves.
Immunohistochemical Studies of DNA Damage Repair Foci
3µm paraffin-embedded tissue sections were softened on a heat block and deparaffinized (3 x 10’ in xyline, 2 x 3’ in 100% ethanol, 2 x 3’ in 95% ethanol, and 1 x 3’ in 70% ethanol followed by washing in distilled water). Antigen retrieval was performed in boiled 0.1 citric acid buffer (pH 6.0) in a steamer at 125 °C for 5’, and allowed to depressurize and cool to room temperature. Slides were then washed in distilled water, incubated in 0.1% PBS-Triton for 30’, and blocked in a solution of 10% natural goat serum and 2% bovine serum albumin in PBS-Triton. Thereafter, slides, incubated overnight with primary Abs at 4 °C to stain DNA repair foci (see below), were washed 5 x 5’ in PBS-Triton on a shaker, incubated in goat anti-mouse or anti-rabbit IgG (H+L) cross-adsorbed secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, A-11070 and A-11017; 2 mg/ml, dilution 1:400) for 1 h at room temperature, and washed with 1 x 5’ in PBS-Triton and 4 x 5’ in PBS on a shaker. Primary antibodies used to stain DNA repair proteins are as follows: mouse monoclonal anti-γH2AX-phosphoSer139 (Milli-pore [clone JBW301] 05-636; dilution 1:1000), rabbit polyclonal anti-BRCA1 (Ser1387) (Novus Biologicals, NB100-225SS; dilution 1:250), and mouse monoclonal anti-DNA-PKcs (phospho-Thr2609) (Abcam, ab18356; dilution 1:250). Slides were mounted in ThermoFisher ProLong™ Diamond Antifade Mountant with DAPI and sealed with nail polish before imaging.
Microscopy for Foci Imaging
Multi-channel fluorescence snapshots of slides were acquired with a Zeiss Axio2 Imaging Microscope with AxioCam MRm Camera (1376 x 1105 pixel images) and the 40x Objective EC “Plan-Neofluar” 100x/1.3 Oil Pol M27 objective. Exposure time for the 488 nm channel for foci wavelengths (marked by Alexa Fluor 488) was calculated to obtain intermediate intensity and avoid oversaturation of foci, maintaining consistency within experiments. Exposure time for 365 nm channel for nucleus wavelengths (marked by DAPI) was calculated according to intermediate intensity without oversaturation. Images were taken from 6 regions from each tumor sample for focus analysis.
Quantification of Foci
Focus microscopic images were evaluated in ImageJ. Numbers of foci were determined by selecting regions of interest corresponding to individual nuclei, calculating mean fluorescence area per nucleus and dividing the area by 8.28 in accordance with published data from our lab on average focus size [10].
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (version 8.4.2 for Windows; San Diego, USA). Values represent mean and standard error of the mean (SEM) as indicated. P-values were calculated using the unpaired two-tailed Student’s t-test with 95% confidence intervals.
Results
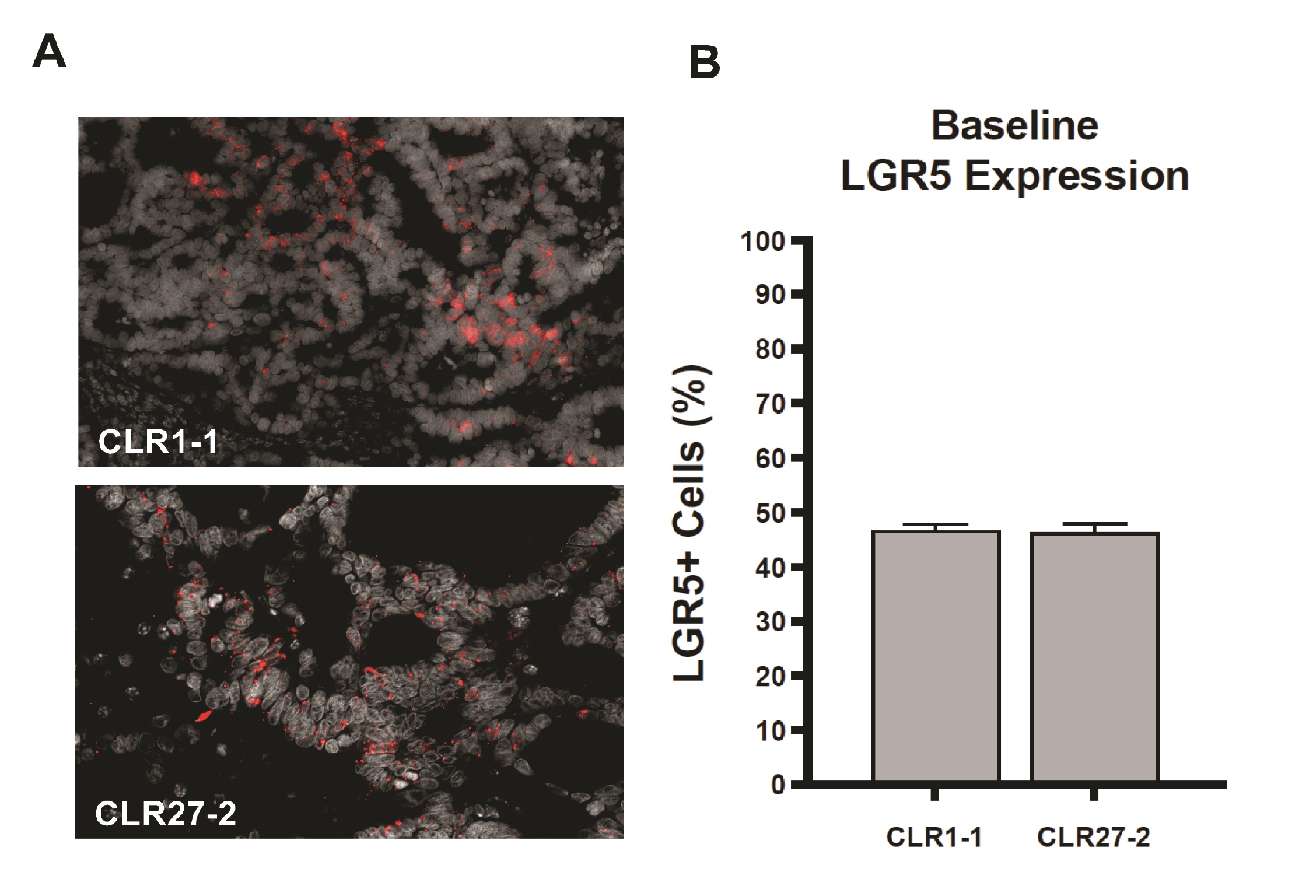
Initial studies determined whether the CRC CLR1-1 and CLR27-2 patient-derived xenografts (PDXs) studied here maintained the logarithmic increase in Lgr5-expressing cells routinely reported for CRC specimens obtained at surgery. Whereas normal human colon displays 3-5% Lgr5+ stem cells at the crypt base [4, 23] both CLR1-1 and CLR27-2 display 40-50% Lgr5+ cells as determined by in situ hybridization (ISH) staining (Fig. 2A-B). In prior studies of normal mouse small and large intestines in situ, in normal colonic organoids cultured ex vivo, and in symmetrically-dividing stem cell colonies ex vivo, we previously provided evidence that sensitivity of the Lgr5+ intestinal stem cell compartment determined outcome of treatment with a single dose radiation exposure [3, 4]. Here we examine whether Lgr5+-enriched PDXs display a similar phenotype.
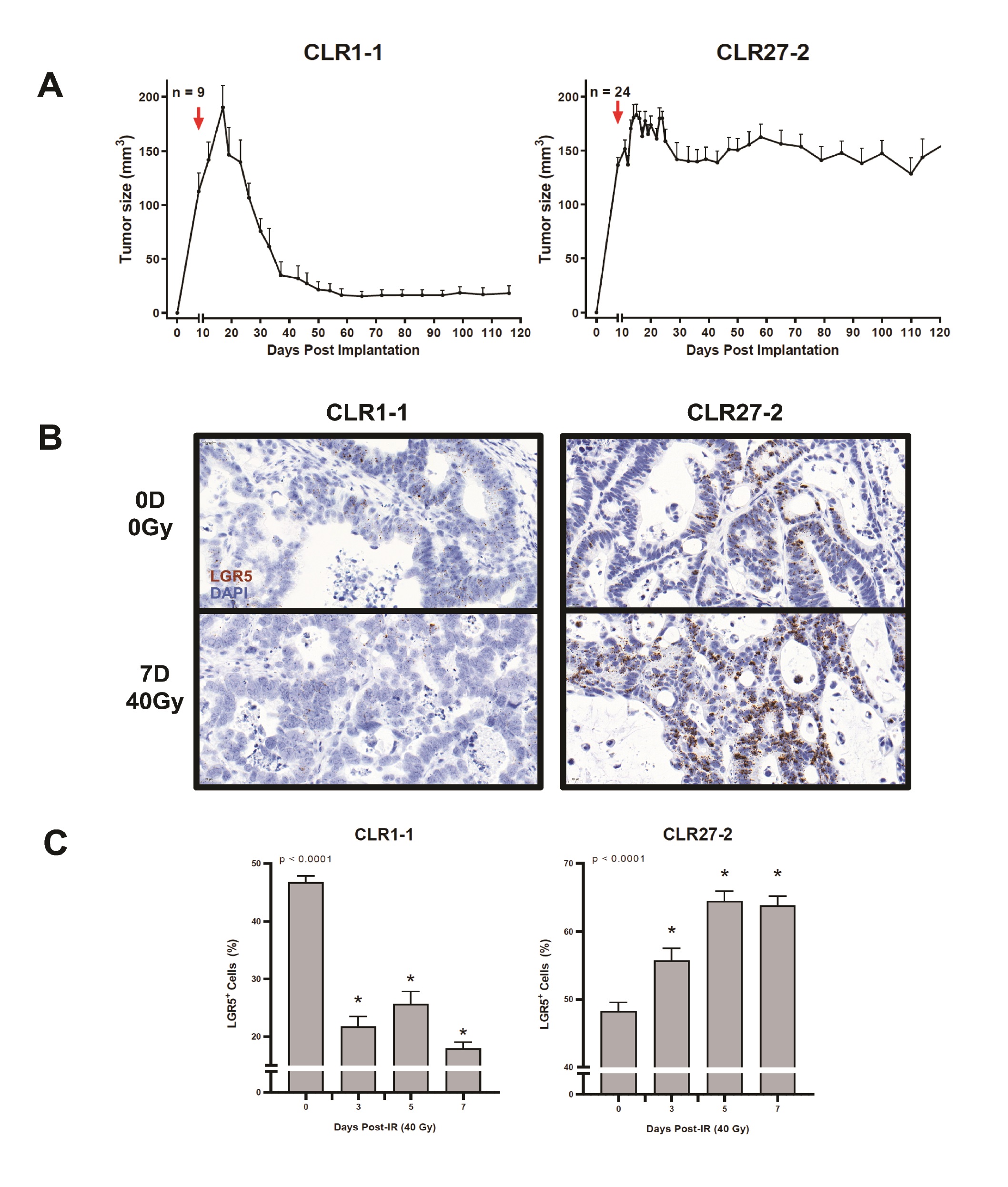
Fig. 3A shows that despite similar percentages of tumor Lgr5+ stem cell-like cells in CLR1-1 and CLR27-2 tumors, these tumors were found to be differentially sensitive to SDRT, with CLR1-1 being sensitive to 40Gy, whereas CLR27-2 was radiation resistant. Furthermore, Fig. 3B-C show that 7 days post 40Gy the radiosensitive CLR-1 PDXs display a marked loss of Lgr5+ stem cells, whereas radioresistant CLR-27-2 tumors show increased Lgr5+ staining. Whereas loss of Lgr5+ stem in CLR1-1 reaches a nadir by day 3 post irradiation, which is maintained for at least 7 days, the radioresistant CLR27-1 shows progressive increase in the Lgr5+ stem cell compartment that peaks at 5 days post radiation and returns to the pre-radiation level by day 14 (Supplementary Fig. 1 – for all supplementary material see www.cellphysiolbiochem.com). Note, CLR1-1 and CLR27-2 Lgr5 ISH show qualitative differences in ISH staining. CLR1-1 tumor cells have large nuclei and display light-colored stippled Lgr5+ foci, while CLR27- 2 display small nuclei and dark dense foci, creating the misperception there might be unequal numbers of positive cells between the two lines. Although the reason for these differences is unknown, close inspection and quantitation reveals similar percentages of positive nuclei in both lines at baseline. These studies indicate that a radiosensitive CRC preferentially deletes stem cells in the immediate post-SDRT period, while the radioresistant variant preferentially deletes non-stem cells, resulting in a rapid increase in the relative proportion of the Lgr5+ stem cell compartment.
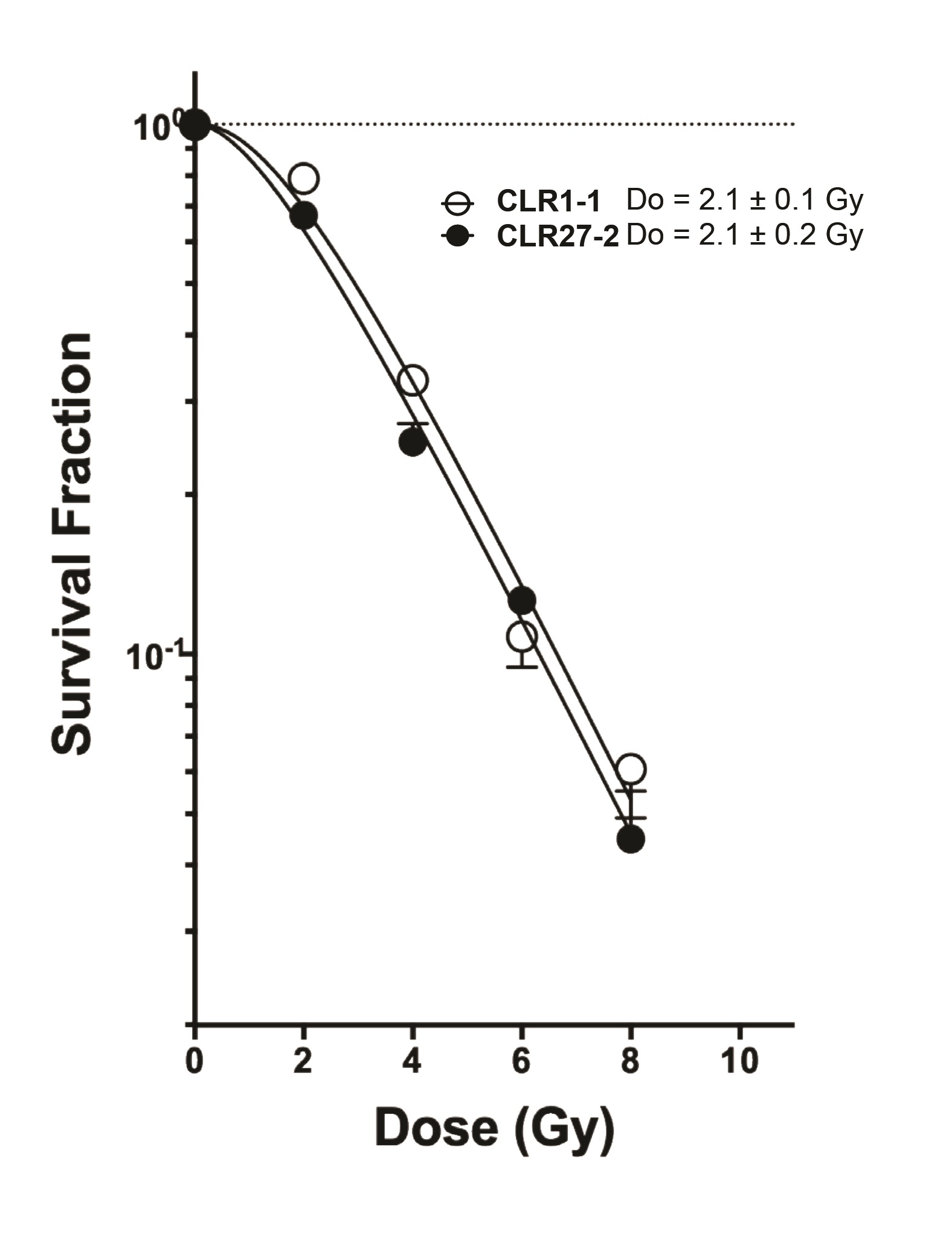
A tenet of conventional radiobiology is that differences in tumor radiosensitivity reflect differences in the inherent DSB repair capacity of tumor parenchymal clonogens [24-27], while the radiation response of the tumor microenvironment plays an insignificant role in treatment outcome [28]. To directly address this concept relative to the observed radiosensitivities of the CLR1-1 and CLR27-2 CRC xenografts, we profiled the respective radiosensitivity phenotypes when grown as organoids ex vivo , using a cutting edge quantitative radiobiologic technique recently published by us for this purpose [3, 18, 29]. The experimental method uses the single hit multi-target (SHMT) algorithm to calculate the radiosensitivity index, originally developed to transform radiation clonogenic cell survival curves by dose into a single numerical value [30]. We adapted this technology to evaluate organoid dose survival curves fitted by nonlinear regression, and transformed by the SHMT algorithm to yield a single D0 value that serves as a numerical estimate of the efficiency of DSB repair [3]. Notably, the higher the D0 value, the greater the radioresistance. Surprisingly, Fig. 4 shows that the radiosensitive CLR1-1 tumor organoids and the radioresistant CLR27-2 tumor organoids show similar radiosensitive profiles, as indicated by identical D0 values of 2.1 Gy. These studies defy an accepted dogma in the field, strongly implying that resistance of CLR27-2 to SDRT is determined by an interaction between tumor parenchymal cells and tumor microenvironment, as described below.
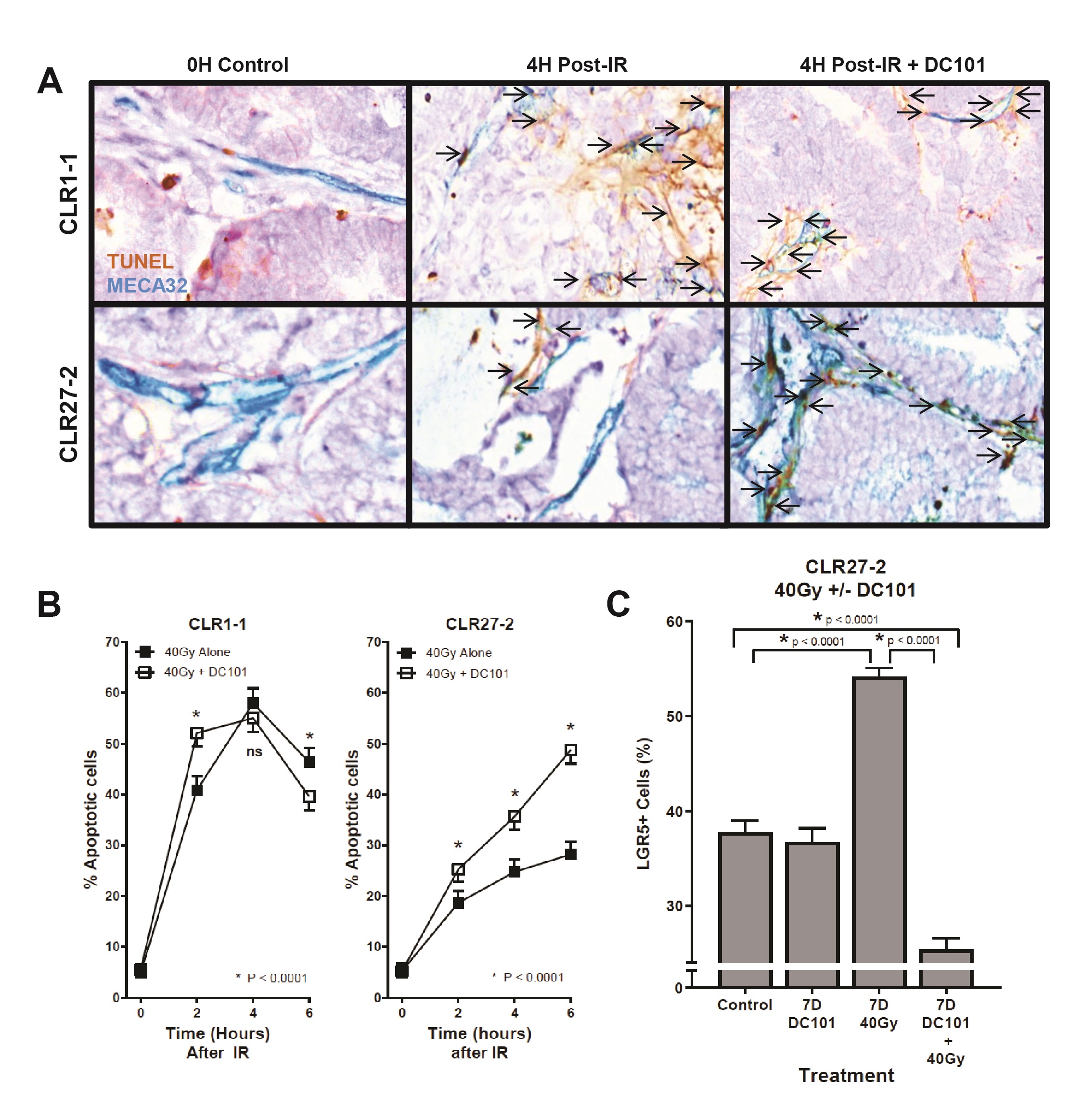
Our prior studies on the high curative impact of SDRT compared with conventional fractionated radiotherapy indicate that SDRT cure results from coupling of acute microvascular injury to parenchymal tumor cell DSB repair, rendering synthetic lethality of tumor clonogens [10]. The data showed that SDRT injury to endothelia renders vasoconstriction by 30 min followed by endothelial apoptosis at 4 h. We initially examined impact of SDRT on the endothelial compartment of radiosensitive CLR1-1 and radioresistant CLR27-2 PDXs. Fig. 5A shows representative images of the CLR1-1 and CLR27-2 PDXs, double-stained with MECA32 and TUNEL to identify apoptotic endothelial cells at 4h post 40Gy, the time of maximal apoptosis induction, quantified in Fig. 5B. At baseline, both tumors display 5-6% apoptotic endothelium that increases to a maximum 58±2% in CLR1-1, but only to 28±2% in CLR27-2. These studies correlate CRC microvascular radiosensitivity with Lgr5+ stem cell content post radiation and tumor response.
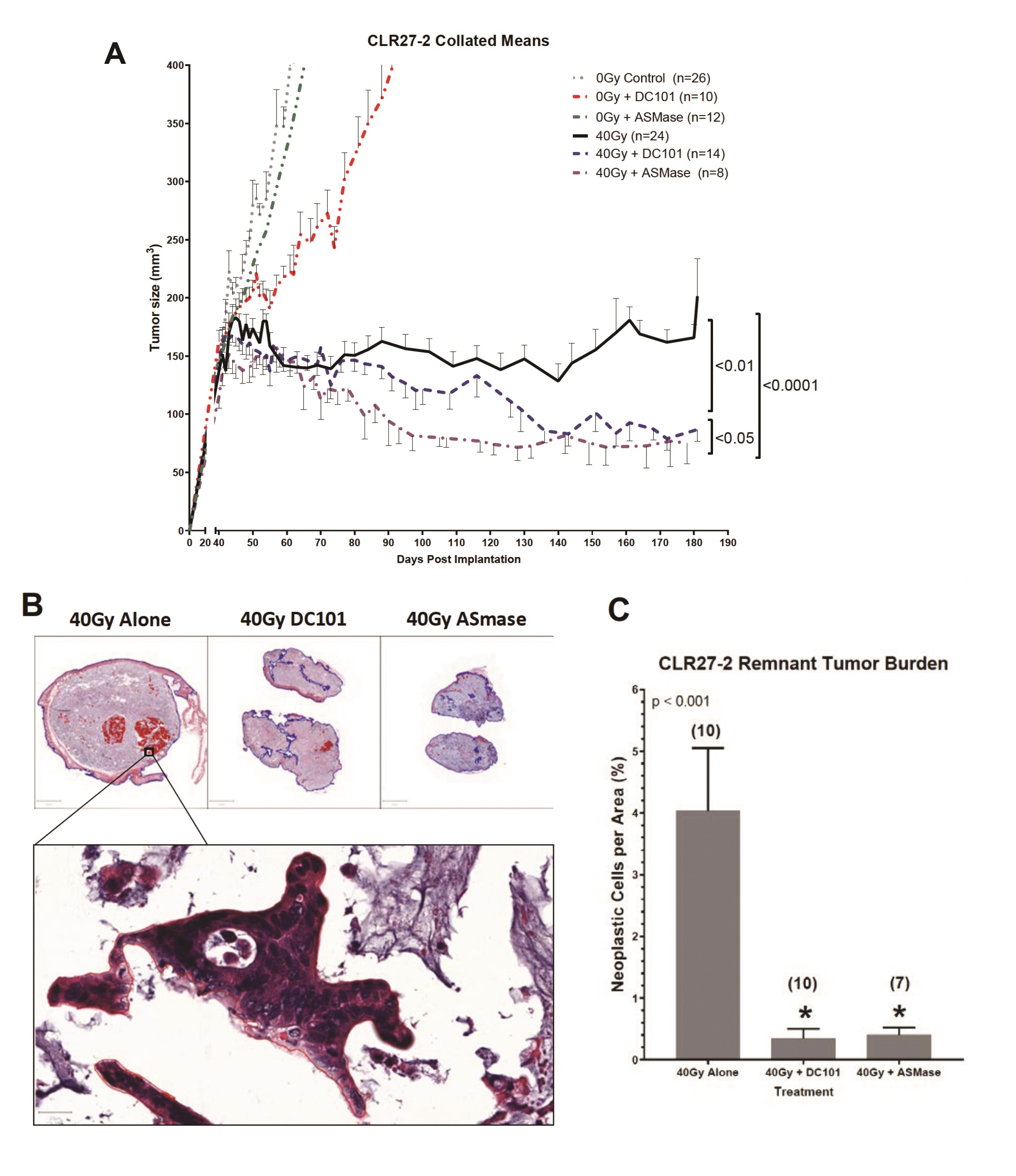
Whereas we published that tumor VEGF inhibits SDRT-induced ASMase activation thereby abrogating neoangiogenic tumor endothelial injury and SDRT lethality in diverse mammalian solid tumors implanted in mice [21, 31, 32], but not in normal mouse tissue, we examined impact of enhancing ASMase/ceramide signaling on CRC PDX responses. Our previous studies showed that anti-angiogenic drugs, when provided at 1h preceding SDRT, optimally de-repress ASMase and enhance SDRT-induced vascular injury [31, 32]. Fig. 5B shows that the VEGFR2 inhibitor DC101 [the murine parental Ab for the clinically-approved Cyramza (ramucirumab)] does not impact SDRT-induced endothelial apoptotic death in the already radiosensitive CLR1-1 PDX, but markedly enhances endothelial cell apoptosis in the radioresistant CLR27-2 PDX into the range associated with effective tumor response in CLR1-1. Further, DC101 prevents the relative concentration of radioresistant Lgr5+ stem cells at 7 days post radiation in CLR27-2, and in fact results, as in CLR1-1 PDXs, in preferential loss of Lgr5+ stem cells (Fig. 5C). Consistent with these findings, DC101 applied at 1h before 40Gy SDRT yields a statistically significant reduction in tumor mass as compared with 40Gy alone (Fig. 6A). As human residual tumor masses often display scar tissue that takes prolonged periods of time to resolve, we collaborated with A.M., a Board Certified Veterinary Pathologist in the Comparative Animal Core Facility at MSKCC, to define residual tumor cells contained within the tumor mass. Fig. 6B shows that there is a significantly larger residual tumor mass in CLR27-2 PDXs treated with 40Gy alone at 130 days post radiation, as compared with tumor residua in mice harboring CLR27-2 PDXs that were pre-treated with DC101 at 1h preceding 40Gy SDRT. Quantitation of this residual tumor mass indicates 20-fold reduction in CLR27-2 tumor cells with DC101 pre-treatment (p<0.001; Fig. 6C).
A complementary approach to increase ASMase/ceramide signaling was employed to confirm that enhancing SDRT-induced ASMase signaling determines loss of Lgr5+CRC stem cells. For these studies, we employed a strategy previously reported by us [19] to overexpress human ASMase exclusively in tumor neo-angiogenic vasculature using adenoviral delivery of a construct that contains a pre-pro endothelin promoter only active in dividing endothelial cells. Notably, >99% of normal tissue endothelial cells in mice and humans are in G0, and hence do not transcribe ASMase from our vector [20]. We show here that delivery of 1x1010 PFU of H2E-PPE1(3x)-ASMase at 5 days preceding 40Gy SDRT resulted, as with timed delivery of DC101, in converting the SDRT-induced increase in Lgr5+ CRC stem cells at 7d post radiation to a selective loss of the Lgr5+ population (Supplementary Fig. 2). Consistent with Lgr5+ cells determining overall CLR27-2 PDX response, combination of ASMase gene therapy with SDRT yielded statistically significant reduction in tumor mass that upon direct pathological analysis revealed a 20-fold reduction in residual tumor cells (Fig. 6). Hence, two distinct approaches to increase ASMase signaling of vascular dysfunction, timed delivery of an anti-angiogenic drug to optimize ASMase activation and ASMase overexpression, similarly increased SDRT-induced CRC stem cell loss and tumor response in radioresistant human CRC PDX CLR27-2.
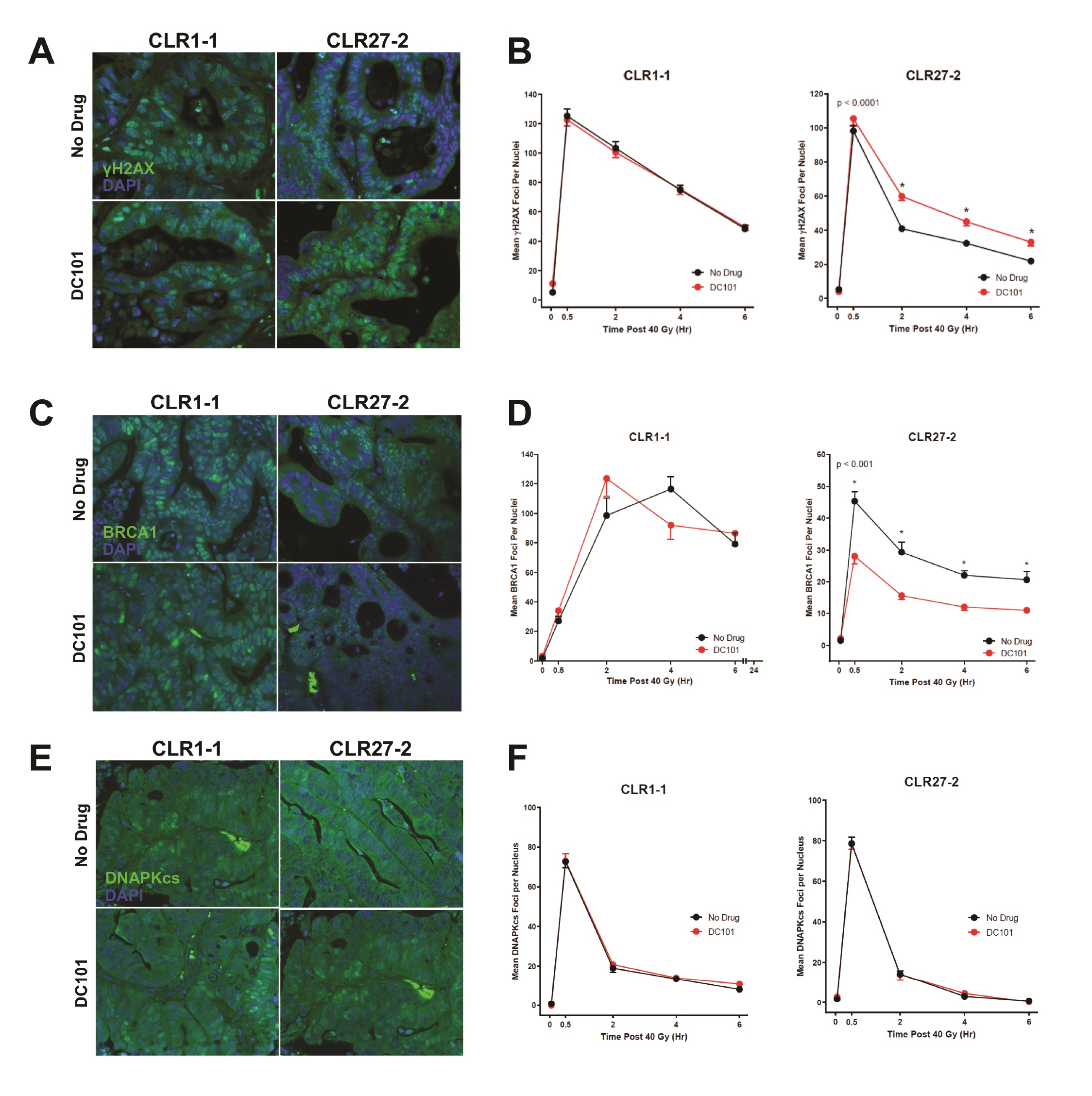
Whereas our prior studies identified murine adult Lgr5+ intestinal stem cells as preferentially using HRR to repair DSBs compared with their progeny [4], and in view of the evidence indicating that SDRT-induced vascular dysfunction inactivates, via the SSR, multiple HRR enzymes as part of tumor cure [10], we examined impact of DC101 ASMase/ceramide radiosensitization on the DNA damage response (DDR). For these studies, we tested the hypothesis that anti-VEGF radiosensitization might specifically attenuate HRR activation following SDRT in CLR27-2, as determined by DNA repair focus technology. Initial investigations quantified kinetics of γH2AX repair foci, the biomarker of choice for assessing global DSB repair in vivo [33], pretreating CRC PDX tumor-bearing mice with or without DC101 (1.6mg DC101/25 gm mouse) at 1h before 40Gy SDRT. By consensus accrual of γH2AX foci, which peaks 0.5-1.0h post irradiation in mammalian tissue, is considered to reflect induction of DSBs, whereas resolution of γH2AX foci represents repair of DSBs. Whereas minimal foci were detected prior to irradiation, Fig. 7A, B (left panel) show thats at 0.5h CLR1-1 and CLR27-2 tumors display 100-120 γH2AX foci/nucleus, respectively, with or without anti-angiogenic drug pre-treatment. Hence, DC101 does not impact accrual of DSB damage consistent with the well-described effect of radiation injury as representing biophysical damage to DNA [34]. In contrast, resolution of DNA damage differs between these tumors with DC101 having no impact on radiosensitive CLR1-1 DSB repair (Fig. 7B, left panel), consistent with its lack of effect on endothelial injury, while the CLR27-2 tumors showed delayed resolution of γH2AX foci, displaying 30-50% more unrepaired DSB damage at all time points (Fig. 7B, right panel). To examine the mechanism of reduction of DSB repair, subsequent studies examined accrual and resolution of BRCA1 and DNA-PKcs foci, established measures of HRR and NHEJ, respectively. Fig. 7C, D shows that while DC101 pre-treatment had no impact on loading of BRCA1 onto damaged DNA, required for optimal HRR, in radiosensitive CLR1-1 PDXs, DC101 pre-treatment specifically attenuated BRCA1 focus formation at 0.5-6 h in radioresistant CLR27-2 PDXs, reducing its contribution to HRR management of DSB repair. Consistent with SDRT impacting HRR specifically, there was no impact of DC101 on NHEJ post SDRT, as measured by focus accrual and resolution of the critical NHEJ mediator of DSB repair, DNA-PKcs (Fig. 7E, F).
We thank Yevgeniy Romin for assistance in confocal imaging.
Authors Contributions
CL, JF, and JC performed most of these studies based on methodology implemented by YP and CL. SB performed focus staining experiments, CL and SK performed ISH investigations, and RF stained tissue for endothelial apoptosis, which was quantified by JC. Histology staining was by RF, while pathologic analysis was by CL with supervision from AM. RK and JF provided critical insight in experimental design and data interpretation. ZZ performed statistical analysis on the data. CL, RK, and ZF jointly prepared the manuscript with input from all authors. ZF and RK conceptualized and supervised the work.
Funding
Funding was provided by NIH grant R01 CA158367 (to RK), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, The Center for Experimental Therapeutics at Memorial Sloan Kettering Cancer, the Virginia and D.K. Ludwig Fund for Cancer Research (to ZF), and the Larry and Stephanie Flinn Foundation (to ZF). This research was funded in part through the NIH/NCI Cancer Center Support Core Grant P30 CA008748 (to RK).
Patents unrelated to this work: RK (US7195775B1, US7850984B2, US10052387B2, US10414533B2, US20140205543A1), RK/JAR (US8562993B2, US9592238B2, US10450385B2, US20150216971A1, US20170335014A1, US20190389970A1, US20190046538A1), RK/ZF (US20170333413A1, US20180015183A1), ZF (US10413533B2). RK/ZF are co-founders of Ceramedix Holding L.L.C. The remaining authors have declared that no conflicts of interest exist.
| 1 Deng X, Michaelson D, Tchieu J, Cheng J, Rothenstein D, Feldman R, Lee SG, Fuller J, Haimovitz-Friedman A, Studer L, Powell S, Fuks Z, Hubbard EJ, Kolesnick R: Targeting Homologous Recombination in Notch-Driven C. elegans Stem Cell and Human Tumors. PLoS One 2015;10:e0127862. https://doi.org/10.1371/journal.pone.0127862 |
||||
| 2 Hua G, Wang C, Pan Y, Zeng Z, Lee SG, Martin ML, Haimovitz-Friedman A, Fuks Z, Paty PB, Kolesnick R: Distinct Levels of Radioresistance in Lgr5(+) Colonic Epithelial Stem Cells versus Lgr5(+) Small Intestinal Stem Cells. Cancer Res 2017;77:2124-2133. https://doi.org/10.1158/0008-5472.CAN-15-2870 |
||||
| 3 Martin ML, Adileh M, Hsu KS, Hua G, Lee SG, Li C, Fuller JD, Rotolo JA, Bodo S, Klingler S, Haimovitz-Friedman A, Deasy JO, Fuks Z, Paty PB, Kolesnick RN: Organoids Reveal That Inherent Radiosensitivity of Small and Large Intestinal Stem Cells Determines Organ Sensitivity. Cancer Res 2019;80:1219-1227. https://doi.org/10.1158/0008-5472.CAN-19-0312 |
||||
| 4 Hua G, Thin TH, Feldman R, Haimovitz-Friedman A, Clevers H, Fuks Z, Kolesnick R: Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology 2012;143:1266-1276. https://doi.org/10.1053/j.gastro.2012.07.106 |
||||
| 5 Greco C, Pares O, Pimentel N, Louro V, Santiago I, Vieira S, Stroom J, Mateus D, Soares A, Marques J, Freitas E, Coelho G, Seixas M, Lopez-Beltran A, Fuks Z: Safety and Efficacy of Virtual Prostatectomy With Single-Dose Radiotherapy in Patients With Intermediate-Risk Prostate Cancer: Results From the PROSINT Phase 2 Randomized Clinical Trial. JAMA Oncol 2021;7:700-708. https://doi.org/10.1001/jamaoncol.2021.0039 |
||||
| 6 Greco C, Kolesnick R, Fuks Z: Conformal Avoidance of Normal Organs at Risk by Perfusion-Modulated Dose Sculpting in Tumor Single-Dose Radiation Therapy. Int J Radiat Oncol Biol Phys 2021;109:288-297. https://doi.org/10.1016/j.ijrobp.2020.08.017 |
||||
| 7 Zelefsky MJ, Yamada Y, Greco C, Lis E, Schoder H, Lobaugh S, Zhang Z, Braunstein S, Bilsky MH, Powell SN, Kolesnick R, Fuks Z: Phase 3 Multi-Center, Prospective, Randomized Trial Comparing Single-Dose 24 Gy Radiation Therapy to a 3-Fraction SBRT Regimen in the Treatment of Oligometastatic Cancer. Int J Radiat Oncol Biol Phys 2021;110:672-679. https://doi.org/10.1016/j.ijrobp.2021.01.004 |
||||
| 8 Greco C, Pares O, Pimentel N, Louro V, Morales J, Nunes B, Castanheira J, Oliveira C, Silva A, Vaz S, Costa D, Zelefsky M, Kolesnick R, Fuks Z: Phenotype-Oriented Ablation of Oligometastatic Cancer with Single Dose Radiation Therapy. Int J Radiat Oncol Biol Phys 2019;104:593-603. https://doi.org/10.1016/j.ijrobp.2019.02.033 |
||||
| 9 Greco C, Fuks Z: Forging New Strategies in the Cure of Human Oligometastatic Cancer. JAMA Oncol 2020;6:659-660. https://doi.org/10.1001/jamaoncol.2020.0195 |
||||
| 10 Bodo S, Campagne C, Thin TH, Higginson DS, Vargas HA, Hua G, Fuller JD, Ackerstaff E, Russell J, Zhang Z, Klingler S, Cho H, Kaag MG, Mazaheri Y, Rimner A, Manova-Todorova K, Epel B, Zatcky J, Cleary CR, Rao SS, et al.: Single-dose radiotherapy disables tumor cell homologous recombination via ischemia/reperfusion injury. J Clin Invest 2019;129:786-801. https://doi.org/10.1172/JCI97631 |
||||
| 11 Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z, Gulbins E, Kolesnick R: Ceramide enables fas to cap and kill. J Biol Chem 2001;276:23954-23961. https://doi.org/10.1074/jbc.M101866200 |
||||
| 12 Stancevic B, Kolesnick R: Ceramide-rich platforms in transmembrane signaling. FEBS Lett 2010;584:1728-1740. https://doi.org/10.1016/j.febslet.2010.02.026 |
||||
| 13 Li PL, Zhang Y, Yi F: Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid Redox Signal 2007;9:1457-1470. https://doi.org/10.1089/ars.2007.1667 |
||||
| 14 Zhang AY, Yi F, Zhang G, Gulbins E, Li PL: Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006;47:74-80. https://doi.org/10.1161/01.HYP.0000196727.53300.62 |
||||
| 15 Alonso D, Radomski MW: The nitric oxide-endothelin-1 connection. Heart Fail Rev 2003;8:107-115. https://doi.org/10.1023/A:1022155206928 |
||||
| 16 Lewicki MC, Srikumar T, Johnson E, Raught B: The S. cerevisiae SUMO stress response is a conjugation-deconjugation cycle that targets the transcription machinery. J Proteomics 2015;118:39-48. https://doi.org/10.1016/j.jprot.2014.11.012 |
||||
| 17 Enserink JM: Sumo and the cellular stress response. Cell Div 2015;10:4. https://doi.org/10.1186/s13008-015-0010-1 |
||||
| 18 Martin ML, Zeng Z, Adileh M, Jacobo A, Li C, Vakiani E, Hua G, Zhang L, Haimovitz-Friedman A, Fuks Z, Kolesnick R, Paty PB: Logarithmic expansion of LGR5(+) cells in human colorectal cancer. Cell Signal 2018;42:97-105. https://doi.org/10.1016/j.cellsig.2017.09.018 |
||||
| 19 Kim JH, Alfieri AA, Kim SH, Young CW: Potentiation of radiation effects on two murine tumors by lonidamine. Cancer Res 1986;46:1120-1123. | ||||
| 20 Stancevic B, Varda-Bloom N, Cheng J, Fuller JD, Rotolo JA, Garcia-Barros M, Feldman R, Rao S, Weichselbaum RR, Harats D, Haimovitz-Friedman A, Fuks Z, Sadelain M, Kolesnick R: Adenoviral transduction of human acid sphingomyelinase into neo-angiogenic endothelium radiosensitizes tumor cure. PLoS One 2013;8:e69025. https://doi.org/10.1371/journal.pone.0069025 |
||||
| 21 Cheng J, Fuller J, Feldman R, Tap W, Owa T, Fuks Z, Kolesnick R: Enhancement of Soft Tissue Sarcoma Response to Gemcitabine through Timed Administration of a Short-Acting Anti-Angiogenic Agent. Cell Physiol Biochem 2020;54:707-718. https://doi.org/10.33594/000000250 |
||||
| 22 Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z, Kolesnick R: Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003;300:1155-1159. https://doi.org/10.1126/science.1082504 |
||||
| 23 Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H: Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003-U1001. https://doi.org/10.1038/nature06196 |
||||
| 24 Foray N, Arlett CF, Malaise EP: Radiation-induced DNA double-strand breaks and the radiosensitivity of human cells: a closer look. Biochimie 1997;79:567-575. https://doi.org/10.1016/S0300-9084(97)82005-6 |
||||
| 25 Jackson SP, Bartek J: The DNA-damage response in human biology and disease. Nature 2009;461:1071-1078. https://doi.org/10.1038/nature08467 |
||||
| 26 Withers HR: Failla memorial lecture. Contrarian concepts in the progress of radiotherapy. Radiat Res 1989;119:395-412. https://doi.org/10.2307/3577512 |
||||
| 27 Rothkamm K, Lobrich M: Misrepair of radiation-induced DNA double-strand breaks and its relevance for tumorigenesis and cancer treatment (review). Int J Oncol 2002;21:433-440. https://doi.org/10.3892/ijo.21.2.433 |
||||
| 28 Budach W, Taghian A, Freeman J, Gioioso D, Suit HD: Impact of stromal sensitivity on radiation response of tumors. J Natl Cancer Inst 1993;85:988-993. https://doi.org/10.1093/jnci/85.12.988 |
||||
| 29 Muschel RJ, Hammond EM, Dewhirst MW: A New Assay to Measure Intestinal Crypt Survival after Irradiation: Challenges and Opportunities. Cancer Res 2020;80:927-928. https://doi.org/10.1158/0008-5472.CAN-19-4045 |
||||
| 30 Hall EJ, Giaccia AJ: Radiobiology for the Radiologist. Philadelphia, Lippincott Williams & Wilkin, 2018, 8th ed. | ||||
| 31 Rao SS, Thompson C, Cheng J, Haimovitz-Friedman A, Powell SN, Fuks Z, Kolesnick RN: Axitinib sensitization of high Single Dose Radiotherapy. Radiother Oncol 2014;111:88-93. https://doi.org/10.1016/j.radonc.2014.02.010 |
||||
| 32 Truman JP, Gueven N, Lavin M, Leibel S, Kolesnick R, Fuks Z, Haimovitz-Friedman A: Down-regulation of ATM protein sensitizes human prostate cancer cells to radiation-induced apoptosis. J Biol Chem 2005;280:23262-23272. https://doi.org/10.1074/jbc.M503701200 |
||||
| 33 Goodarzi AA, Jeggo PA: Irradiation induced foci (IRIF) as a biomarker for radiosensitivity. Mutat Res 2012;736:39-47. https://doi.org/10.1016/j.mrfmmm.2011.05.017 |
||||
| 34 Ward JF: The yield of DNA double-strand breaks produced intracellularly by ionizing radiation: a review. Int J Radiat Biol 1990;57:1141-1150. https://doi.org/10.1080/09553009014551251 |
||||
| 35 Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN: Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756-760. https://doi.org/10.1038/nature05236 |
||||
| 36 Glumac PM, LeBeau AM: The role of CD133 in cancer: a concise review. Clin Transl Med 2018;7:18. https://doi.org/10.1186/s40169-018-0198-1 |
||||