×
![]()
Corresponding Author: Michela Terlizzi
Department of Pharmacy, DIFARMA, University of Salerno
Via Giovanni Paolo II 132, Fisciano, 84084, Salerno (Italy)
E-Mail mterlizzi@unisa.it
Intracellular Sphingosine-1-Phosphate Receptor 3 Contributes to Lung Tumor Cell Proliferation
Michela Terlizzia Chiara Colarussoa Giusy Ferraroa Maria Chiara Montia Ilaria De Rosab Pasquale Sommab Rosario Salvic Aldo Pintoa Rosalinda Sorrentinoa
aUniversity of Salerno, Department of Pharmacy, DIFARMA, Fisciano, Italy, bAnatomy and Pathology Unit, Ospedale dei Colli, AORN, “Monaldi”, Naples, Italy, cThoracic Surgery Unit, Ospedale dei Colli, AORN, “Monaldi”, Naples, Italy
Introduction
Recent evidence has highlighted the relevance of the axis sphingosine/sphingosine-1-phosphate (S1P) in both physiological and pathological conditions [1-4]. Sphingosine can derive from both the activation of sphingomyelinase and/or serine-palmitoyltransferase [5]. The sphingomyelinase is an enzyme which induces membrane sphingolipids degradation to generate ceramide, which in turn is converted by the ceramidase into sphingosine. The alternative pathway for sphingosine synthesis is related to the serine-palmitoyltransferase which promotes the so-called de novo synthesis through the condensation of serine to palmitoyl-CoA [6]. Both metabolic pathways lead to ceramide generation that on one side could be transformed into ceramide-1-phosphate (C1P) and, on the other lead to the formation of sphingosine, which will be phosphorylated by sphingosine kinases I and/or II (SPHK I and/or SPHK II). Once S1P is generated, it is transported through the cell membrane in an inside-out mode by means of Spinster homolog 2 (SPNS2), a non-ATP-dependent organic ion transporter [7], so that it can interact in a paracrine and/or autocrine manner with its receptors, sphingosine-1-phosphate receptors (S1PRs). So far, five G protein-coupled S1PRs have been identified, S1PR1, S1PR2, S1PR3, S1PR4 and S1PR5, which are involved in physiological and pathological conditions, including cell growth, apoptosis, trafficking, differentiation and activation of immune cells, inflammation, cellular architecture and vascular integrity regulation [8-11].
So far, S1PR1, a Gi-coupled receptor, and S1PR3, a Gi/q-coupled receptor, have been demonstrated as involved in tumor growth. In particular, S1PR1 is largely considered as a pro-tumorigenic receptor in that it promotes neo-angiogenesis [12], metastasis of various cancers (i.e., breast and colorectal cancer, thyroid carcinoma), via the activation of STAT3, extracellular signal–regulated kinase (ERK), Akt, and Rac pathways [13]. Similarly, S1P/S1PR3 axis is associated with proliferation, migration and angiogenesis of cancer cells [14] via the activation of TGF-β/SMAD3 axis [15]. Little is known about S1PR2 and S1PR4, although it looks like that their activity is majorly on immune cells [16, 17].
In our recent study we found that S1P supported Toll-like Receptor (TLR)-9/NF-κB-mediated TNF-α release from lung adenocarcinoma cells, but not from non-pathological cells, mainly via the inside-out transport of TLR-9-induced S1P which interacted with S1PR3 [18]. In support, epidemiological studies have revealed that higher levels of S1P are detected in patients with chronic respiratory disorders, but in particular, in lung cancer patients [19, 20].
Based on our previous study in which we found that S1P/S1PR3 is involved in an inflammatory pattern via the activation of TLR-9, highly expressed on lung cancer cells, the main goal of the current study was to better understand the involvement of S1P/S1PR3 pathway/signaling during lung carcinogenesis, taking advantage of a mouse model of first-hand smoke exposure and of a carcinogen-induced lung cancer. We found that the intranuclear, but not the membrane, localization of S1PR3 was associated to the proliferation of lung adenocarcinoma cells, mechanism that was correlated to human and mouse samples of smoke-exposure and carcinogen-induced lung cancer, which were characterized by higher utilization of S1P.
Materials and Methods
Human tissues
Samples human tissues were obtained by patients with diagnosis of Non-Small Cell Lung Cancer (NSCLC) or non-neoplastic pulmonary pathologies (i.e tuberculosis, emphysema) who underwent surgical resection at Ospedale dei Colli, AORN, Monaldi, Naples, Italy, during the period 2014-2017. Clinical data, lifestyle and histology reports were obtained from the Pathological Anatomy Unit of the hospital. The project was approved by the institutional review board and by the Ethical Committee (approval number for lung cancer patients 1254/2014). Samples from lung cancer and non-cancerous patients were collected after oral and written information provided by the MDs, and after the signature of a consent form before entering the project. Samples were collected and used within 24 hours. Lung cancer patients were 60±10 (mean±S.E.M.) years of age.
Mice
Female specific pathogen-free C57Bl/6N mice (6-8 weeks of age) (Charles River Laboratories, Lecco, Italy) were fed with a standard chow diet and maintained in specific pathogen-free conditions at the animal care facility of Department of Pharmacy, University of Salerno. This study was carried out in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the Health National Institute. The experimental protocol was approved by the Ethical Committee for Animal Studies of the University of Salerno and Health Ministry with the approval number 985/2017. All animal experiments were performed under protocols that followed the Italian (D.L. 26/2014) and European Community Council for Animal Care (2010/63/EU). Experiments were repeated twice. Each group was composed of 9 mice.
Cigarette smoke mice-exposure protocol
As previously reported [21], mice were exposed to first-hand smoking by means of a nose-only chamber (EMMS, UK). Briefly, mice were exposed to first-hand smoking once a day at the concentration of 1 μg/cm3 of total particulate matter (TPM), generated from Red Marlboro cigarettes (TAR = 12 mg/cig), 5 days/week for 8 and 16 weeks. Control mice, here defined as Room Air group, breathed filtered air for the same time. Mice were sacrificed 24 hours after the last cigarette smoke (CS) exposure.
Carcinogen-induced lung cancer in mice
Mice were anesthetized with isoflurane and intratracheally (i.t.) instilled with Benzo(a)pyrene (BaP) (SUPELCO Analytical, 595 North Harrison Road, Bellefonte, PA, USA; Cat#40071) at the dose of 1 µg/mouse in 10 µl at week 1, 2, 3, week 8-9-10 and week 12-13-14. Vehicle group (acetone 0.1%; Sigma-Aldrich, 3050 Spruce Street, St. Louis, MO, USA; Cat#32201) was considered. Lungs were isolated and left lung lobes were embedded into OCT medium to perform H&E staining as described below. Right lung lobes were digested with 1U/mL collagenase (Sigma Aldrich, 3050 Spruce Street, St. Louis, MO, USA; Cat#C0130). Cell suspensions were passed through 70 μm cell strainers, and red blood cells were lysed. Cell suspensions were used for flow cytometric analyses of different cell subtypes. Lungs were homogenized and western blotting were performed. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Salerno and by National Institutes of Health with the approval number 683/2019.
Tumor lesion evaluation
H&E staining was performed to evaluate tumor lesion development after carcinogen exposure. Tumor lesion was expressed as ratio of Tumor Area/Total Lung Area, calculated with ImageJ software (NIH, USA).
Flow Cytometry Analysis
Cell suspensions obtained by collagenase digested lungs were analysed using flow cytometry (BD FacsCalibur, Milan, Italy) to evaluate the infiltration and the nature of immune cells recruited to the lung of mice. Cell suspensions were labelled with specific antibodies (CD11b: 8999 BioLegend Way, San Diego, CA; Cat#101239; Gr-1: 8999 BioLegend Way, San Diego, CA; Cat#108408; Arginase-1: Proteintech, 5400 Pearl Street, Rosemont, IL, USA; Cat#16001-1-AP; CD3: 8999 BioLegend Way, San Diego, CA; Cat#100322; CD4: 8999 BioLegend Way, San Diego, CA; Cat#130308; CD25: 8999 BioLegend Way, San Diego, CA; Cat#101904; FoxP3: Thermo Fisher Scientific, 3747 Meridian Road, Rockford, IL, USA; Cat#PA5-12396).
Western blotting analysis
Human and mouse lung homogenates were used to examine the expression of S1PR1 (Santa Cruz Biotechnologies, Heidelberg, Germany; Cat#sc-48356), S1PR3 (Alomone labs, Jerusalem, Israel; Cat#ASR-013), SPNS2 (Abcam, Milan, Italy; Cat#ab82629), ASAH1 (N-acylsphingosine amidohydrolase 1, ASAH1 or ceramidase; Elabscience; US; Cat#E-AB-10959). GAPDH (OriGene Technologies, Rockville, MD; Cat#TA890003) and HSC70 (OriGene Technologies, Rockville, MD; Cat#TA332519) were used as loading protein. Data were analysed by means of ImageJ software (NIH, USA).
Immunofluorescence analyses
Mice and human lung cryosections were analysed for the presence of FITC-conjugated S1PR3. DAPI (4’,6-diamidino-2-phenylindole) was used to stain cell nuclei. Images were observed by means of Carl Zeiss confocal microscopy (magnification: 40X).
Sphingolipids extraction from mouse tissues
Extraction of sphingolipids, sphingosine (S) and sphingosine-1-phosphate (S1P), from lung homogenates was performed by the following method [22]. Briefly, 1ml of mixture i–PrOH–Water–EtOAc (30:10:60) was added to 0.2 mg of total proteins in each lung tissue homogenate. Samples were stirred for 30 minutes, sonicated for 15 minutes and centrifuged at 21000 g for 15 minutes. The supernatant was transferred to a new centrifuge tube and evaporated to dryness at 40°C in the vacuum rotator. Samples were reconstituted in 50 µl MeOH, vortexed for 5 minutes, sonicated for 10 minutes and centrifuged for 10 minutes at 10000 g and 8 µl were loaded on the UPLC-MSMS system.
LC-MS/MS analysis
Sphingosine and S1P were quantified by liquid chromatography coupled to tandem mass spectrometry detection (LC-MS/MS) on Shimadzu LC-20A and Auto Sampler systems and QTRAP 6500 instrument from AB-Sciex. C18 chromatographic column (Kinetex C18, 50 x 2.1 mm, 5µm, Phenomenex) was used for chromatographic separation. The mobile phase was composed of water 0.1% formic acid (mobile phase A) and methanol 0.1% formic acid (mobile phase B). The flow rate was set at 400 µl/minute. The mass spectrometer was set in the positive ion mode (ESI+) with an electrospray voltage of 5500 V at 400°C of the heated capillary temperature. The multiple reaction monitoring (MRM) mode and the Analyst 1.6.2 software has been used. Sphingosine was analyzed with the mass transition 300 m/z to 282 m/z and it was observed at the rt of 6.65 minutes. S1P was analyzed with the mass transition 380 m/z to 264 m/z and it was observed at the rt of 7.95 minutes. Nitrogen was used as the air curtain gas (20psi), atomizing gas (30psi), auxiliary gas (60psi) and collision gas (4psi). Dwell time was 100ms, DP was 74v, EP was 10v, CE was 22v and CXP was 15v. For quantitative analysis, a standard curve with sphingosine and S1P amounts of 250-2.5 pg has been run. Data were expressed as sphingosine and S1P Peak Area.
Proliferation assay
A549 cells (ATCC® CCL-185™) were stained by means of carboxyfluorescein diacetate succinimidyl ester (CFSE; 1µM; proliferation Dye, eBioscience Inc., San Diego, CA; Cat#65-0850-85) to perform proliferation assay. CFSE flow cytometry data was analysed by means of ModFit4.0 software (BD Pharmingen). A549 cells were treated for 48 hours with sphingosine-1-phosphate (S1P 10 nM; Sigma-Aldrich, Merck Life Science S.r.l., Milan, Italy; Cat#S9666) or unmethylated Cytosine phosphate Guanosine-oligodeoxynucleotides, CpG-ODN (CpG 1 μg/ml; Vincibiochem, Milan, Italy) in presence of specific pharmacological inhibitors, such as TY52156, a S1PR3 receptor antagonist (S1PR3a 10 µM; Tocris Bioscience, Ellisville, MO; Cat#5328), anti-Spinster homolog 2 antibody (αSPNS2 10 ng/ml; Abcam, Milan, Italy; Cat#ab82629), SKI II, a selective inhibitor of sphingosine kinases (SKI II 10 µM; Tocris Bioscience, Ellisville, MO; Cat#2097).
Nuclear and cytosolic protein extraction
Nuclear and cytosolic protein were isolated from A549 cells, after 3 hours treatment with CpG 1 μg/ml, by using Qproteome Nuclear Protein Kit and Qproteome Plasma Membrane Protein Kit according to the manufacturer’s instructions (QIAGEN, United Kingdom). S1PR3 and SPHK II expression was evaluated in the different cellular compartments by means western blotting analysis. GAPDH was used as cytosolic loading protein and Lamin A (Sigma-Aldrich, Merck Life Science S.r.l., Milan, Italy; Cat#L1293) as nuclear loading protein.
Statistical Analysis
Data are reported as median ± percentile range and represented as dot plots. Statistical differences were assessed with TWO-WAY or ONE-WAY Analysis of variance (ANOVA) followed by multiple comparison post-tests as appropriate. p values less than 0.05 were considered significant. The statistical analysis was performed by using GraphPad prism 9.0.0 version (San Diego, USA).
Results
Sphingosine-dependent pathway in human samples of NSCLC
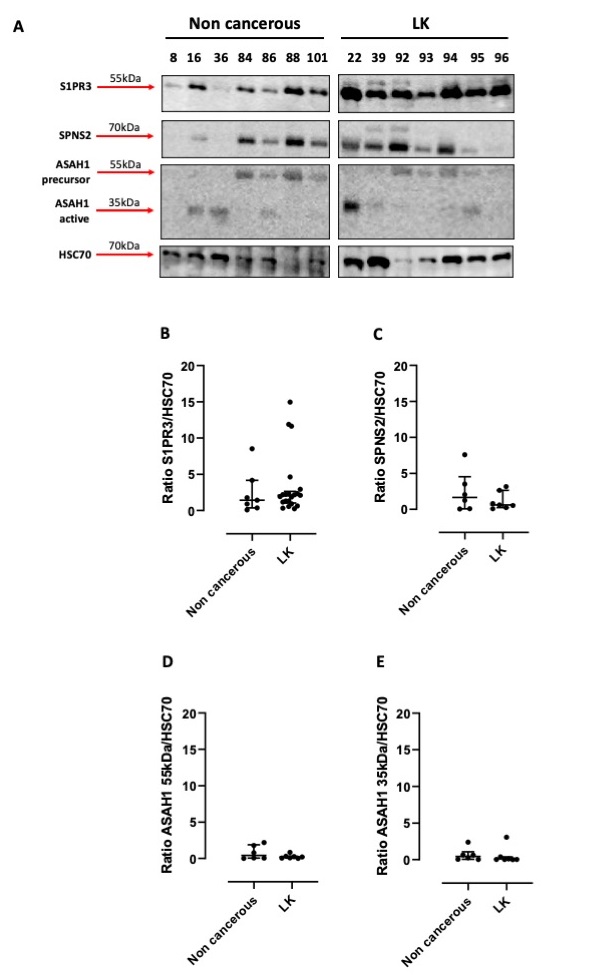
In our previous study we found that the activation of sphingosine/S1P axis in human tumor cells was associated to the release of a pro-inflammatory cytokine, TNF-α [18]. This effect was not observed in non-cancerous cells (i.e., healthy human fibroblasts), implying that this mechanism was effective in the sole tumor cells. In order to understand whether this pathway was involved in lung carcinogenesis, we used human tissues of NSCLC and non-cancerous patients undergoing thoracic surgery. We found that S1PR3 was expressed in all samples of NSCLC (Fig. 1A). Similarly, it was also detectable in non-cancerous lung samples (Fig. 1A), although at a different content. However, the quantitative analysis performed by ImageJ software did not show statistically differences of S1PR3 expression in cancerous (LK) vs non-cancerous lung tissue homogenates (Fig. 1A and 1B). Instead, we were not able to detect S1PR1 (data not shown).
The expression of the inside-out S1P transporter, SPNS2, was not altered in tumor tissues (LK) compared to non-cancerous tissues (Fig. 1A and 1C). Moreover, we did not find any statistical difference in both non-cancerous and tumor samples in the expression of ceramidase (ASAH1), the enzyme involved in sphingosine synthesis (Fig. 1A, 1D and 1E). However, we have to point out the active form of the ceramidase (ASAH1, 35kDa) was evident in 2 cases of tuberculosis (patient 16 and 36) and in a patient (lane #84) who had fibrosis (Fig. 1A, 1E).
These data may suggest that sphingosine-dependent signaling is similar in lung tumor cells as well as in non-cancerous cells.
S1P pathway in lung samples of smoking mice
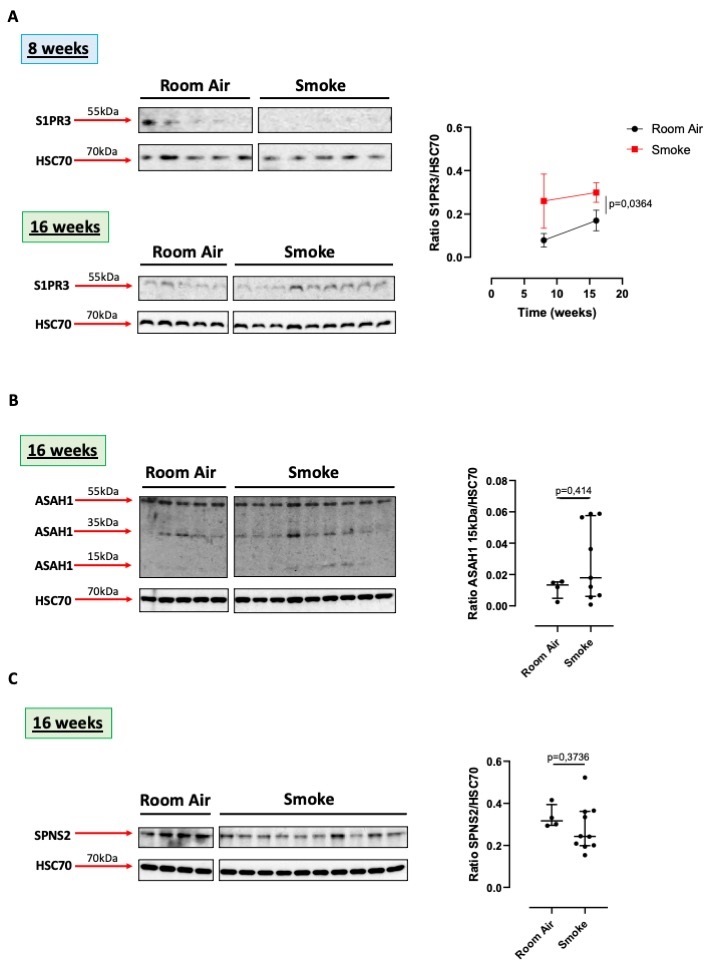
Smoking represents nowadays the high-risk factor that can induce lung cancer. Therefore, we used a model of first-hand smoking mice to evaluate whether smoking-associated lung inflammation, was correlated to sphingosine-dependent pathway. We used lung homogenates of mice which were sacrificed at 8 and 16 weeks, time points that revealed altered alveolar structure, collagen deposition, inducing an emphysematous pattern typical of chronic obstructive pulmonary disease (COPD) [21]. In these experimental conditions, S1PR3 was higher expressed in the lung of smoking mice at 16 weeks compared to 8 weeks and to the relative control group (Room Air) (Fig. 2A). Again, we were not able to detect S1PR1 (data not shown).
To evaluate whether sphingosine was produced starting from the metabolism of the ceramide, we first analyzed the expression of the ceramidase (ASAH1). The precursor form of the acid ceramidase (55kDa) was similarly expressed in the lung of Room Air and smoking group of mice exposed for 16 weeks (Fig. 2B). Instead, the smaller proteic fragments (35 and 15 kDa), representing the active form of the enzyme, seemed (not in a statistical manner) to be higher in the lung of smoking mice than in the Room Air group (Fig. 2B).
Interestingly, the expression of SPNS2 was higher in the lung of the Room Air group than in mice exposed for 16 weeks to smoke (Fig. 2C).
Therefore, the presence of smaller fragments of the ceramidase, 15 and 35 kDa, may imply the formation of S1P following smoking exposure. However, this effect was not correlated to the expression of SPNS2, S1P inside-out transporter, suggesting that smoke-induced S1P holds an intracellular signal rather than an extracellular/autocrine/paracrine signal.
S1P pathway in a mouse model of carcinogen-induced lung cancer
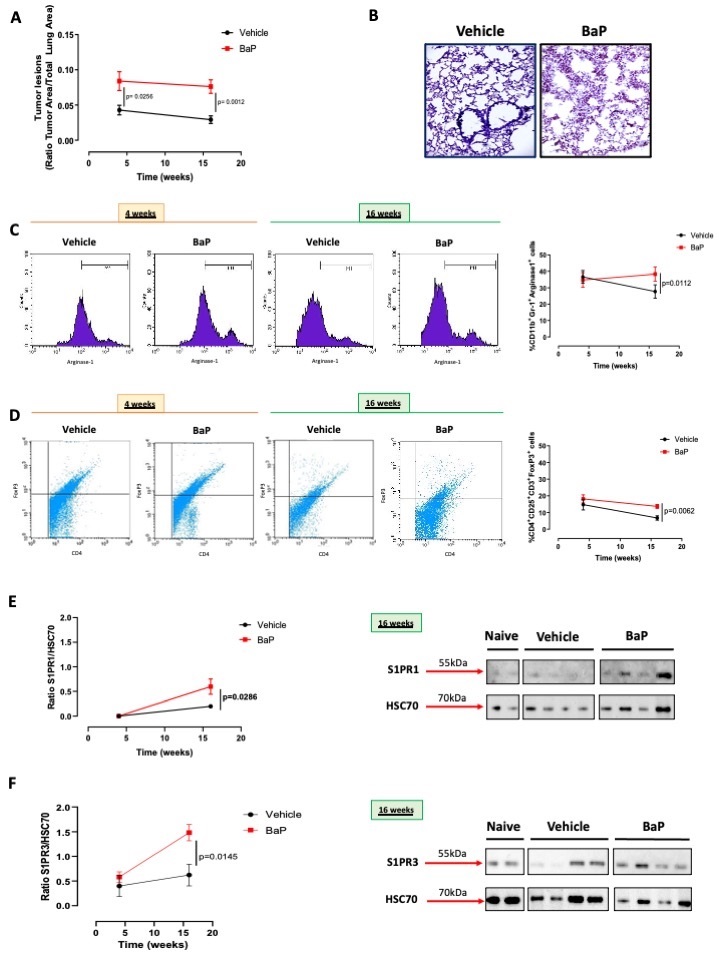
Benzo(a)pyrene (BaP) is one of the major components of tobacco smoking that has carcinogen-like activities. Therefore, in order to understand the role of the sphingosine/S1P axis in a tobacco-induced lung cancer, we used BaP that was instilled into C57Bl/6 mice intratracheally, according to the experimental protocol described in the Material and Method section. The administration of BaP induced the formation of tumor lesions into the lung of mice compared to the vehicle group (acetone 0.1%) (Fig. 3A and 3B). The lung of BaP-treated mice was characterized by the presence of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs: identified as CD11b+Gr-1+Arginase-1+) (Fig. 3C) and T regulatory cells (Treg: identified as CD3+CD4+CD25+FoxP3+) (Fig. 3D). Both the percentage of MDSCs (Fig. 3C) and Treg (Fig. 3D) were statically higher in BaP-treated mice than vehicle mice.
The expression of S1PR1 at 16 weeks was significantly higher in the lung of BaP-treated mice (Fig. 3E). Similarly, the expression of S1PR3 at 16 weeks was higher in the carcinogen-induced lung tumor model than the vehicle group (Fig. 3F). Because the vehicle group was treated with acetone 0.1%, we also analyzed naive (non-treated mice) as comparison (Fig. 3E and 3F).
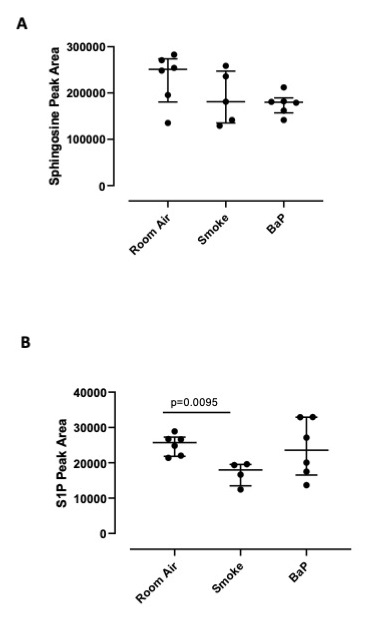
To prove that sphingosine was phosphorylated, we used lung homogenates of Room Air, smoking- and BaP-treated mice and performed LC-MS/MS experiments. We found that the lung of Room Air mice had the highest content of sphingosine (Fig. 4A), which did not differ in terms of Peak Area than S1P, implying a physiological role of sphingosine/S1P in the lung, as already reported [3]. Instead, the lung of smoking and BaP-treated mice had lower levels of sphingosine (Fig. 4A), which corresponded to lower levels of S1P in the sole smoking mice but not in BaP-treated mice (Fig. 4B), implying that smoking induces the degradation of S1P and most likely its cellular activity is exerted, as already reported [23]. In support, the lung of smoking mice had higher active ceramidase (35 and 15 kDa, Fig. 2B) than Room Air mice, which leads to suggest that sphingosine is metabolized by ceramidase and then phosphorylated to S1P, active metabolite that interacts with its receptors [10].
The intracellular S1P pathway leads to lung tumor cell proliferation
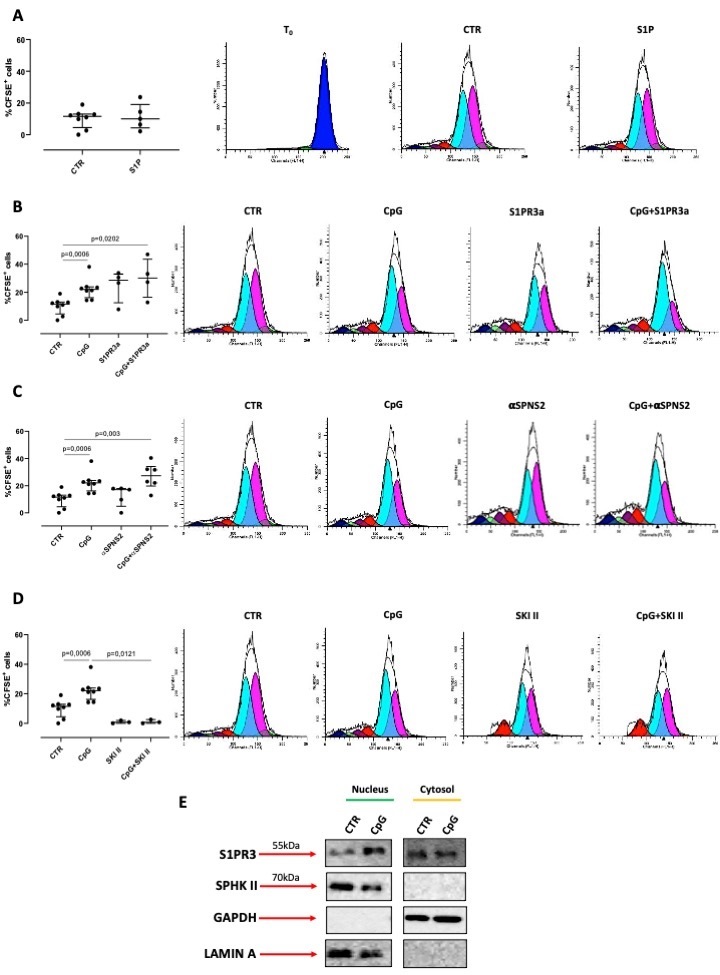
In order to evaluate the molecular mechanism that binds sphingosine/S1P axis to lung carcinogenesis, we used a much simpler experimental approach by using lung adenocarcinoma cells, A549. In order to evaluate whether S1P was able to induce tumor cell proliferation, we treated A549 cells with exogenous S1P. Surprisingly, we found that the stimulation of A549 cells with exogenous S1P (10 nM) was not able to induce A549 cells to proliferate (Fig. 5A). Therefore, we used CpG, a TLR9-ligand, that we previously demonstrated as involved in the release of endogenous S1P [18]. The stimulation of cells with CpG induced A549 cells to proliferate (Fig. 5B). To prove the role of S1P in this context and according to the inside-out signaling of S1P, we blocked S1PR3, with a potent and selective receptor antagonist (S1PR3a). Interestingly and in support to what observed with the exogenous S1P (Fig. 5A), the pharmacological blockade of the extracellular S1PR3 did not alter A549 cell proliferation after CpG treatment (Fig. 5B). Similarly, the blockade of SPNS2 did not statistically modify the CpG-increased percentage of CFSE+ cells (Fig. 5C). In sharp contrast, the inhibition of SPHK I/II, by means of SKI II inhibitor, responsible for sphingosine phosphorylation, completely reduced A549 proliferation after CpG treatment (Fig. 5D). Therefore, our next hypothesis was to understand the intracellular activity of the endogenous S1P.
Interestingly, we found that S1PR3 was also expressed into the nucleus of A549 cells (Fig. 5E). It has to be pointed out that the treatment of cells with CpG increased S1PR3 expression into the nucleus compared to the cytosol/membrane compartment (Fig. 5E). Importantly, we found that SPHK II was present into the nucleus and not in the cytosol/membrane compartments (Fig. 5E), supporting the hypothesis that the pro-tumor activity of S1P is intracellular and more specifically is related to the presence of S1PR3 into the nucleus.
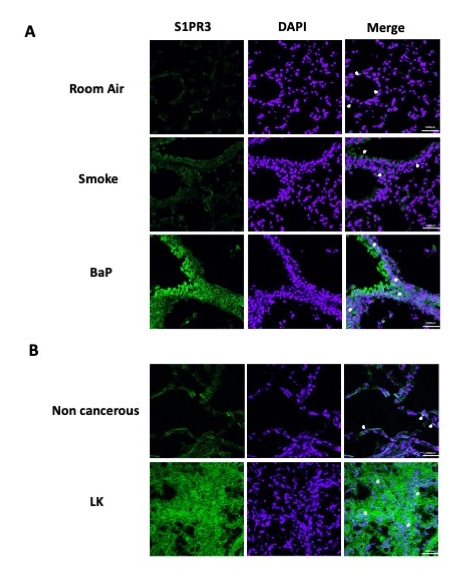
To further prove our hypothesis, we analyzed the expression of S1PR3 in Room Air, smoking and BaP-treated mice by means of fluorescence staining to correlate the in vivo data (Fig. 1, 2 and 3) to the in vitro results (Fig. 5). As hypothesized, S1PR3 was overexpressed in the lung of BaP-treated mice compared to Room Air and smoking groups and it showed both membrane and nuclear localization (Fig. 6A, white arrows). Similarly, human lung cancer tissues expressed both membrane and nuclear S1PR3 compared to non-cancerous tissues (Fig. 6B, white arrows). These data confirm our hypothesis on the role of S1P in lung carcinogenesis via the activation of the nuclear S1PR3.
Author Contributions
MT and CC designed and performed experiments; GF and MCM performed LC-MS/MS data; IDR, PS, RS (Salvi) recruited NSCLC patients; MT, AP and RS (Sorrentino) statistically analyzed and interpreted data and wrote the manuscript.
Funding
This study was supported by FARB-2017-2020 and Italian Association against leukemia (AIL) in favor of RS.
Statement of Ethics
The authors have no ethical conflicts to disclosure. This study was carried out in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the Health National Institute, and was approved by the institutional review board and by the Ethical Committee.
The authors have no conflicts of interest to declare.
| 1 Pyne S, Adams DR, Pyne NJ: Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog Lipid Res 2016;62:93-106. https://doi.org/10.1016/j.plipres.2016.03.001 |
||||
| 2 Roviezzo F, Sorrentino R, Bertolino A, De Gruttola L, Terlizzi M, Pinto A, Napolitano M, Castello G, D'Agostino B, Ianaro A, Sorrentino R, Cirino G: S1P-induced airway smooth muscle hyperresponsiveness and lung inflammation in vivo: molecular and cellular mechanisms. Br J Pharmacol 2015;172:1882-1893. https://doi.org/10.1111/bph.13033 |
||||
| 3 Hannun YA, Obeid LM: Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol 2018;19:175-191. https://doi.org/10.1038/nrm.2017.107 |
||||
| 4 Riboni L, Abdel Hadi L, Navone SE, Guarnaccia L, Campanella R, Marfia G: Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020;9:337. https://doi.org/10.3390/cells9020337 |
||||
| 5 Proia RL, Hla T: Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 2015;125:1379-1387. https://doi.org/10.1172/JCI76369 |
||||
| 6 Gault CR, Obeid LM, Hannun YA: An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 2010;688:1-23. https://doi.org/10.1007/978-1-4419-6741-1_1 |
||||
| 7 Spiegel S, Maczis MA, Maceyka M, Milstien S: New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J Lipid Res 2019;60:484-489. https://doi.org/10.1194/jlr.S091959 |
||||
| 8 Sorrentino R, Bertolino A, Terlizzi M, Iacono VM, Maiolino P, Cirino G, Roviezzo F, Pinto A: B cell depletion increases sphingosine-1-phosphate-dependent airway inflammation in mice. Am J Respir Cell Mol Biol 2015;52:571-583. https://doi.org/10.1165/rcmb.2014-0207OC |
||||
| 9 Roviezzo F, Sorrentino R, Iacono VM, Brancaleone V, Terlizzi M, Riemma MA, Bertolino A, Rossi A, Matteis M, Spaziano G, Pinto A, D'Agostino B, Cirino G: Disodium cromoglycate inhibits asthma-like features induced by sphingosine-1-phosphate. Pharmacol Res 2016;113:626-635. https://doi.org/10.1016/j.phrs.2016.09.014 |
||||
| 10 Brinkmann V: Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther 2007;115:84-105. https://doi.org/10.1016/j.pharmthera.2007.04.006 |
||||
| 11 Cartier A, Leigh T, Liu CH, Hla T: Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc Natl Acad Sci U S A 2020;117:3157-3166. https://doi.org/10.1073/pnas.1906246117 |
||||
| 12 Balaji Ragunathrao VA, Anwar M, Akhter MZ, Chavez A, Mao Y, Natarajan V, Lakshmikanthan S, Chrzanowska-Wodnicka M, Dudek AZ, Claesson-Welsh L, Kitajewski JK, Wary KK, Malik AB, Mehta D: Sphingosine-1-Phosphate Receptor 1 Activity Promotes Tumor Growth by Amplifying VEGF-VEGFR2 Angiogenic Signaling. Cell Rep 2019;29:3472-3487.e4. https://doi.org/10.1016/j.celrep.2019.11.036 |
||||
| 13 Rostami N, Nikkhoo A, Ajjoolabady A, Azizi G, Hojjat-Farsangi M, Ghalamfarsa G, Yousefi B, Yousefi M, Jadidi-Niaragh F: S1PR1 as a Novel Promising Therapeutic Target in Cancer Therapy. Mol Diagn Ther 2019;23:467-487. https://doi.org/10.1007/s40291-019-00401-5 |
||||
| 14 Lee HM, Lo KW, Wei W, Tsao SW, Chung GTY, Ibrahim MH, Dawson CW, Murray PG, Paterson IC, Yap LF: Oncogenic S1P signalling in EBV-associated nasopharyngeal carcinoma activates AKT and promotes cell migration through S1P receptor 3. J Pathol 2017;242:62-72. https://doi.org/10.1002/path.4879 |
||||
| 15 Zhao J, Liu J, Lee JF, Zhang W, Kandouz M, VanHecke GC, Chen S, Ahn YH, Lonardo F, Lee MJ: TGF-β/SMAD3 Pathway Stimulates Sphingosine-1 Phosphate Receptor 3 Expression: IMPLICATION OF SPHINGOSINE-1 PHOSPHATE RECEPTOR 3 IN LUNG ADENOCARCINOMA PROGRESSION. J Biol Chem 2016;291:27343-27353. https://doi.org/10.1074/jbc.M116.740084 |
||||
| 16 Rivera J, Proia RL, Olivera A: The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol 2008;8:753-763. https://doi.org/10.1038/nri2400 |
||||
| 17 Olesch C, Sirait-Fischer E, Berkefeld M, Fink AF, Susen RM, Ritter B, Michels BE, Steinhilber D, Greten FR, Savai R, Takeda K, Brüne B, Weigert A: S1PR4 ablation reduces tumor growth and improves chemotherapy via CD8+ T cell expansion. J Clin Invest 2020;130:5461-5476. https://doi.org/10.1172/JCI136928 |
||||
| 18 Terlizzi M, Colarusso C, Ferraro G, Monti MC, Cerqua I, Roviezzo F, Pinto A, Sorrentino R: Sphingosine-1-Phosphate Contributes to TLR9-Induced TNF-α Release in Lung Tumor Cells. Cell Physiol Biochem 2021;55:222-234. https://doi.org/10.33594/000000361 |
||||
| 19 Milara J, Navarro R, Juan G, Peiró T, Serrano A, Ramón M, Morcillo E, Cortijo J: Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012;67:147-156. https://doi.org/10.1136/thoraxjnl-2011-200026 |
||||
| 20 Alberg AJ, Armeson K, Pierce JS, Bielawski J, Bielawska A, Visvanathan K, Hill EG, Ogretmen B: Plasma sphingolipids and lung cancer: a population-based, nested case-control study. Cancer Epidemiol Biomarkers Prev 2013;22:1374-1382. https://doi.org/10.1158/1055-9965.EPI-12-1424 |
||||
| 21 Colarusso C, Terlizzi M, Lamort AS, Cerqua I, Roviezzo F, Stathopoulos G, Pinto A, Sorrentino R: Caspase-11 and AIM2 inflammasome are involved in smoking-induced COPD and lung adenocarcinoma. Oncotarget 2021;12:1057-1071. https://doi.org/10.18632/oncotarget.27964 |
||||
| 22 Bielawski J, Szulc ZM, Hannun YA, Bielawska A: Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 2006;39:82-91. https://doi.org/10.1016/j.ymeth.2006.05.004 |
||||
| 23 De Cunto G, Brancaleone V, Riemma MA, Cerqua I, Vellecco V, Spaziano G, Cavarra E, Bartalesi B, D'Agostino B, Lungarella G, Cirino G, Lucattelli M, Roviezzo F: Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke-exposed mice. Br J Pharmacol 2020;177:267-281. https://doi.org/10.1111/bph.14861 |
||||
| 24 Hait NC, Maiti A: The Role of Sphingosine-1-Phosphate and Ceramide-1-Phosphate in Inflammation and Cancer. Mediators Inflamm 2017;2017:4806541. https://doi.org/10.1155/2017/4806541 |
||||
| 25 Wang P, Yuan Y, Lin W, Zhong H, Xu K, Qi X: Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int 2019;19:295. https://doi.org/10.1186/s12935-019-1014-8 |
||||
| 26 Maceyka M, Harikumar KB, Milstien S, Spiegel S: Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 2012;22:50-60. https://doi.org/10.1016/j.tcb.2011.09.003 |
||||
| 27 Shu X, Wu W, Mosteller RD, Broek D: Sphingosine kinase mediates vascular endothelial growth factor-induced activation of ras and mitogen-activated protein kinases. Mol Cell Biol 2002;22:7758-7768. https://doi.org/10.1128/MCB.22.22.7758-7768.2002 |
||||
| 28 Olivera A, Rosenfeldt HM, Bektas M, Wang F, Ishii I, Chun J, Milstien S, Spiegel S: Sphingosine kinase type 1 induces G12/13-mediated stress fiber formation, yet promotes growth and survival independent of G protein-coupled receptors. J Biol Chem 2003;278:46452-46460. https://doi.org/10.1074/jbc.M308749200 |
||||
| 29 Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, D'Andrea RJ, Gamble JR, Vadas MA: Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem 2002;277:7996-8003. https://doi.org/10.1074/jbc.M111423200 |
||||
| 30 Wang W, Toran PT, Sabol R, Brown TJ, Barth BM: Epigenetics and Sphingolipid Metabolism in Health and Disease. Int J Biopharm Sci 2018;1:105. https://doi.org/10.31021/ijbs.20181105 |
||||
| 31 di Villa Bianca Rd, Sorrentino R, Sorrentino R, Imbimbo C, Palmieri A, Fusco F, Maggi M, De Palma R, Cirino G, Mirone V: Sphingosine 1-phosphate induces endothelial nitric-oxide synthase activation through phosphorylation in human corpus cavernosum. J Pharmacol Exp Ther 2006;316:703-708. https://doi.org/10.1124/jpet.105.093419 |
||||
| 32 Sorrentino R, Morello S, Giordano MG, Arra C, Maiolino P, Adcock IM, Pinto A: CpG-ODN increases the release of VEGF in a mouse model of lung carcinoma. Int J Cancer 2011;128:2815-2822. https://doi.org/10.1002/ijc.25626 |
||||