×
![]()
Corresponding Author: Catherine Meyer-Schwesinger
Institute of Cellular and Integrative Physiology,
University Medical Center Hamburg-Eppendorf
Martinistraße 52, 20246 Hamburg (Germany)
E-Mail c.meyer-schwesinger@uke.uni-hamburg.de
The Intertwining of Autophagy and the Ubiquitin Proteasome System in Podocyte (Patho)Physiology
Lukas Heintz Catherine Meyer-Schwesinger
Institute of Cellular and Integrative Physiology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Introduction
Our smallest renal filtration unit, the glomerulus comprises four cell types, which form a functional syncytium: The visceral epithelial cells (podocytes), the parietal epithelial cells (PECs) of Bowman’s capsule, the fenestrated and highly glycosylated glomerular endothelial cells (GEnCs), and a type of specialized pericytes termed mesangial cells (MCs) [1]. Podocytes and endothelial cells of the glomerular capillaries are separated by the glomerular basement membrane and form the 3-layered glomerular filtration barrier, over which blood filtration ultimately occurs in a size and charge selective manner. Mesangial cells modulate the size of the filtration area and parietal epithelial cells prevent leakage of the primary urinary filtrate into the tubulointerstitium. Of all glomerular cells, podocytes, as terminally differentiated cells, exhibit the most elaborate morphology with primary processes arising from the podocyte cell body, which form an intricate network of interdigitating foot processes with neighboring podocytes. A specialized form of adherens junction, the slit membrane, interconnects foot processes. With its unique protein repertoire, the slit membrane represents a flexible non-clogging barrier to proteins, with proposed mechano-sensitive properties. Thereby, the slit membrane proteins nephrin and neph1 bridge the slit membrane and podocin generates a signaling hub in lipid rich domains of the podocyte foot process mediating mechano-sensation. The glomerulus can become a target of immune attack by both the humoral and cellular immune system resulting in different forms of glomerulonephritis. Depending on the site of glomerular injury, glomerulonephritis can present as either a nephrotic or as a nephritic syndrome. Nephrotic syndrome is defined by four components: high range proteinuria, hypoalbuminemia, edema, and hypertriglyceridemia and is prototypic for the injury of podocytes. Podocyte injury ensues in a uniform morphological injury pattern combining different degrees of podocyte hypertrophy, podocyte foot process effacement and podocyte loss to the urine, ultimately leading to dialysis necessitating renal insufficiency. As a general theme, these morphological changes appear to involve reactivation of developmental programs such as those engaged by Notch [2], Wnt [3-6], and mTOR pathways [7]. Over-activation, imbalance and impairment of these central intracellular signaling pathways disrupt normal podocyte energy metabolism [8] and protein homeostasis [9] thus initiating a mostly irreversible dedifferentiation process. The significance of proteostatic pathway alterations in podocytes will be the focus of this review, which is based on many excellent expert reviews. We apologize to those many researchers, whose work could not be cited in this context.
Proteostatic systems
The set of proteins, which define the proteome of a given cell, is controlled by a mechanism called proteostasis. Proteostasis comprises cellular processes such as the translation (synthesis) of proteins, the precise folding and localization of proteins following synthesis, and the degradation of flawed and of unneeded proteins by cellular degradation systems. About 240 g protein are synthesized and degraded daily in a 60 kg adult, the majority being intracellular proteins [10]. Failure to accurately regulate these cellular processes ultimately leads to disease. Several degradation systems exist, of which the ubiquitin proteasomal system (UPS) and the autophagosomal lysosomal pathway (ALP) represent the most important systems for intracellular protein degradation. Both systems degrade proteins into small polypeptides and help maintain amino acid pools and energy (ATP) balance either during acute starvation by the UPS or in the course of chronic starvation by autophagy [11] and both are responsible for protein quality control. Thereby, the UPS is responsible for degrading 80-90% of proteins including many regulated, short lived, abnormal, denatured, or in general damaged proteins. Autophagy is primarily responsible for the degradation of most long-lived proteins, but also degrades aggregated proteins as well as cellular organelles.
The Ubiquitin proteasome system
Among the multitude of biological processes regulated by the UPS are the cell cycle, gene transcription and translation, cell survival and apoptosis, cell metabolism and protein quality control, and inflammation through the degradation of specific protein components of these processes. The UPS combines the highly specific mechanisms of 1) substrate tagging and of 2) substrate protein degradation within a multi-protein complex called the proteasome (Fig. 1). Substrate tagging is achieved by the covalent attachment of the conserved protein ubiquitin to one or more lysine residues of the substrate protein via a hierarchical ATP consuming multi-enzymatic process mediated by E1, E2 and E3 enzymes. One single E1 enzyme is responsible for ubiquitin activation. Thereafter, the ubiquitin conjugating E2 enzymes (about 50 known) participate in the transfer of the activated ubiquitin to the protein substrate. In a vast majority of cases, a member of the E3 ubiquitin ligase family also participates in the conjugation process. E3 ligases serve as docking proteins that bring the substrate protein and the E2 carrier protein with the activated ubiquitin together. E3 enzymes are grouped into three major families, namely RING, HECT and RBR families, based on structural similarities and the functional classes of substrates they recognize. Each E3 enzyme recognizes specific proteins destined for ubiquitination, therefore this step is a major mechanism ensuring specificity toward substrate proteins and is performed by over 1000 known E3 ligases. The pattern of ubiquitin conjugation (termed the ubiquitin code) determines the fate of ubiquitinated proteins. Different kinds of ubiquitin linkages can occur, ranging from ubiquitin conjugation to target proteins either as monomers (monoubiquitination) or as di-, oligo- and poly-ubiquitin chains (poly-ubiquitination). Depending on the lysine (K) within the ubiquitin protein used for the generation of polyubiquitin chains, one distinguishes between K6-, K11-, K27-, K29-, K33-, K48-, and K63-polyubiquitin linkages. The level of complexity of the ubiquitin code is enhanced through posttranslational phosphorylation and acetylation of ubiquitin, and by the generation of mixed ubiquitin chains through incorporation of ubiquitin-like modifiers such as SUMO, NEDD8 or FAT10. Deubiquitinating enzymes, furthermore, are able to modify the length and the branching of polyubiquitin chains, thereby altering the fate of the substrate protein. The small number (<100) of DUBs might at first suggest a low degree of selectivity; however, DUBs are subject to multiple layers of regulation that modulate both their activity and their specificity [12]. Four main families of deubiquitinating enzymes are present in mammalian cells: 58 ubiquitinspecific proteases (USPs), 4 ubiquitin C-terminal hydrolases (UCHs), 5 Machado-Josephin domain proteases (MJDs), 14 ovarian tumor proteases (OTU), and Jab1/Mov34/Mpr1 Pad1 N-terminal+ (MPN+) (JAMM) domain-containing genes [13]. Deubiquitinating enzymes cleave ubiquitin from proteins and disassemble polyubiquitin chains that are released from substrates before proteasomal degradation. The activities of deubiquitinating enzymes are important for the recycling of ubiquitin for subsequent ubiquitination reactions, for the prevention of proteasome congestion, and for the control of specific protein turnover by modifying/removing ubiquitin or polyubiquitin chains from the targeted protein and by this changing the fate of the protein, i.e. prevention of proteasomal degradation.
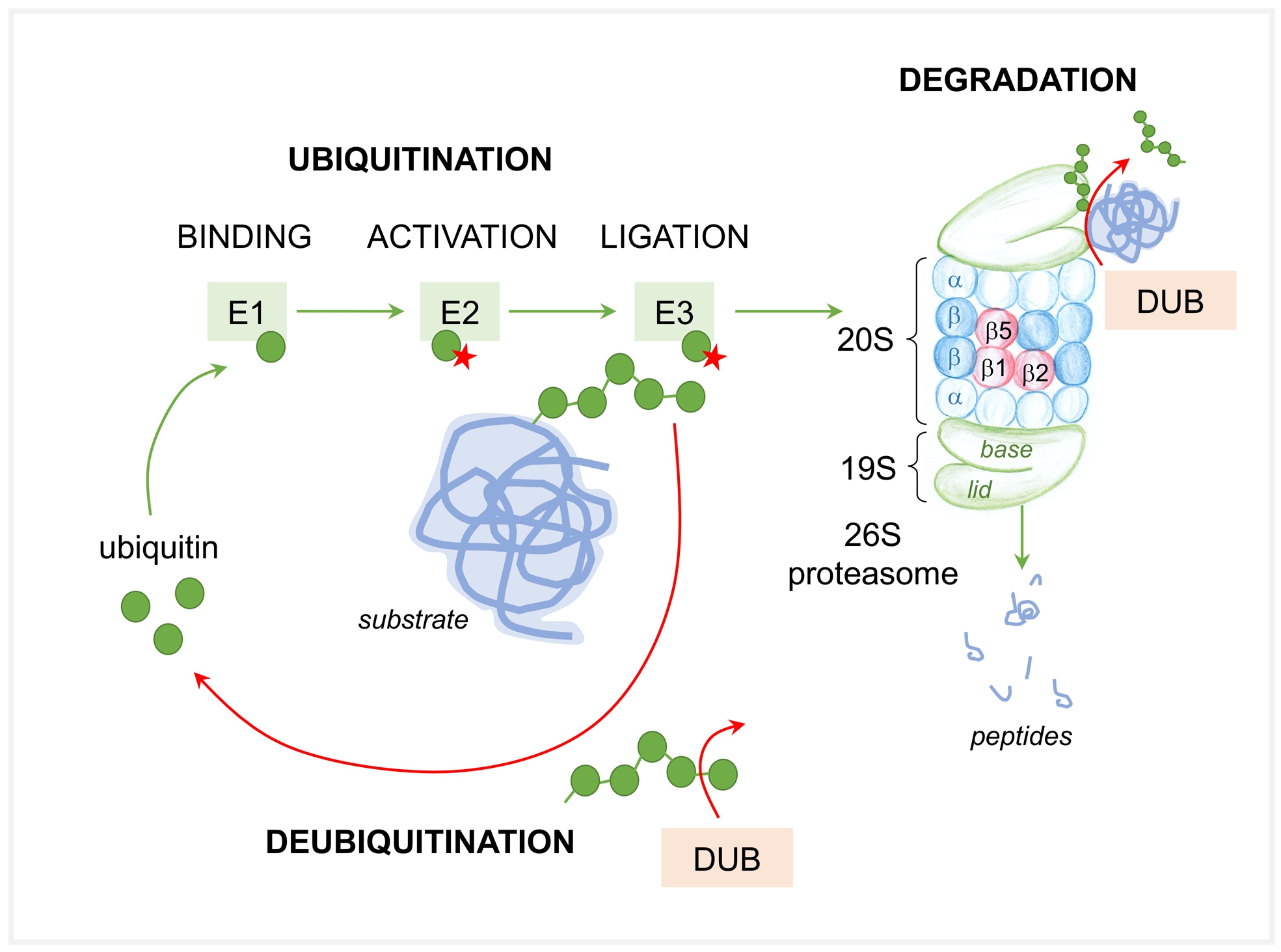
The current concept is that ubiquitination is required for the initiating association of the ubiquitinated proteins with the proteasome complex. This high affinity binding to the proteasome is easily reversible by competition with other ubiquitin binding proteins and does not depend on ATP. Subsequently, the second tight binding step that commits the substrate to proteasome degradation requires ATP hydrolysis and a loosely folded region in the substrate [14]. After ubiquitinated proteins bind to the proteasome complex, proteasomes disassemble and release the ubiquitin chain, engage the deubiquitinated substrate into the proteolytic chamber and degrade the substrate.
The proteasome is a 2.5 MegaDalton protease consisting of over 60 subunits, which are assembled as a hybrid between the 20S catalytic core particle (CP) and the 19S regulatory particle (RP). The 20S CP is a barrel shaped structure of 28 subunits arranged as a cylindrical stack of two outer α-rings and two inner β-rings, composed of 7 structurally similar α or β subunits. The inner two β-rings form the proteolytic chamber, whereas the outer two α-rings serve as a gate for substrate entry into the chamber. The β1, β2, β5 subunits of the constitutive CP harbor the distinct proteolytic activities: The β5 subunit harbors the chymotrypsin-like activity; the β2 subunit the trypsin-like activity; the β1 subunit the caspase-like activity, which degrade the engaged substrates to peptides. The 19S regulatory particle binds to the cylinder end of the CP, and opens a channel located centrally within the cylinder end. The RP contains 19-20 subunits grouped in a base (ATPases) and a lid (non-ATPases), which in a complex interplay mediate substrate binding, substrate deubiquitination, and substrate entry into the proteolytic CP. Of note, the proteasome does not degrade proteins to amino acids, but instead produces a highly heterogeneous mixture of peptides from a given protein [15], which then either serve as raw material for adaptive cell-mediated immunity [16] or which are further processed by cytosolic peptidases to amino acids for de novo protein synthesis. The complexity of the proteasome system is further achieved by the fact that cells can alter their proteolytic capacity and specificity by modulating the amount and diversity of proteasomes. Thus, different types of proteasomes exist based on the types of proteolytic subunits incorporated within the β-rings of the 20S core complex or based on the regulatory caps associated with the 20S core particle. The immunoproteasome (i26S) harbors the β1i, β2i, β5i subunits within its 20S CP, which are thought to mediate a more efficient and / or alternative peptide cleavage ideal for MHC-class 1 presentation [17]. The association of so-called proteasome activators, such as PA28α/β, PA28γ, or PA200, which represent alternative regulatory particles to the 20S CP enhance the open probability of the proteasome and thereby peptide hydrolysis (reviewed in [18]).
The autophagosomal lysosomal pathway
Autophagy is a predominantly nonspecific highly conserved “self-eating” process, which was discovered in the late 1950’s [19]. As one arm of the endocytic system, autophagy is essential for the maintenance of cellular homeostasis and cellular stress response with its main function to provide metabolic precursors for survival in conditions of stress and to serve like the UPS as a protein quality control system by clearing misfolded proteins and other cellular debris. Three types of autophagy are known, which differ in their mechanisms and functions: Microautophagy, chaperone mediated autophagy, and macroautophagy. In microautophagy, small cytoplasmic cargoes are engulfed within lysosomal membrane invaginations [20]. Chaperone mediated autophagy requires the recruitment of KFERQmotive- bearing proteins to the lysosome via binding to chaperones such as heat shock cognate protein 70 (Hsc70) and co-chaperones BAG1, Hip, Hop and Hsp40/DNAJB1 [21].
The KFERQ motive is present in 30% of cellular proteins. The chaperone then binds to a specific lysosomal receptor LAMP-2A on the lysosomal membrane [21], the cargo protein is unfolded and translocated to the lysosome for degradation. Macroautophagy (hereafter termed “autophagy”) is believed to be the major mode of autophagy and is the most described form of autophagy. The principle of macroautophagy is that mainly bulky cargo such as less rapidly degraded proteins, organelles or protein aggregates are removed by engulfing them in a double membrane compartment called the autophagosome, which fuses with lysosomes [22] for final degradation. Lysosomal degradation is highly efficient, as this membrane-enclosed organelle contains numerous proteases of low specificity [23].
Depending on the substrates degraded, macroautophagy is subdivided in mammalian cells into a bulk non-selective form and into selective forms governing the removal of mitochondria (mitophagy), of lipid droplets (lipophagy), of peroxisomes (pexophagy), of ribosomes (ribophagy), of proteasomes (proteophagy), and of lysosomes (lysophagy) [24]. Selectivity of autophagy is controlled by autophagy receptors, which associate simultaneously with cargo and the autophagosomal membrane proteins LC3 (ATG8-or microtubule-associated protein 1A/1B-light chain 3) and its homologous protein GABARAP (γ-aminobutyric acid receptor-associated protein-like). The most studied autophagy receptors are sequestome 1 (SQSTM1)/p62 and neighbor of breast cancer 1 gene (BRCA1) 1, which sequester aggregated proteins to form SQSTM1/p62 bodies for autophagy.
The process of autophagy (Fig. 2) can mechanistically be divided into distinct steps: 1) initiation by cell signaling pathways, 2) nucleation, 3) elongation, and 4) vesicle fusion to lysosomes. The understanding of the molecular mechanisms of autophagy has ensued following the initial discovery of the autophagy related genes (ATG) from genetic studies in yeast [22]. Sixteen ATG proteins comprise the conserved core ATG machinery that catalyzes formation of small vesicles made of a double membrane called phagophore (or isolation membrane) at a specialized site called the phagophore assembly site. In mammals, initiation is associated with an endoplasmic reticulum (ER) subdomain enriched for the lipid phosphatidylinositol 3-phosphate (PI(3)P), known as the omegasome [25]. The origin of the membrane used for phagophore formation may involve different sources, such as Golgi, ER, plasma membrane, recycling endosomes, mitochondria, and lipid droplets [26, 27].
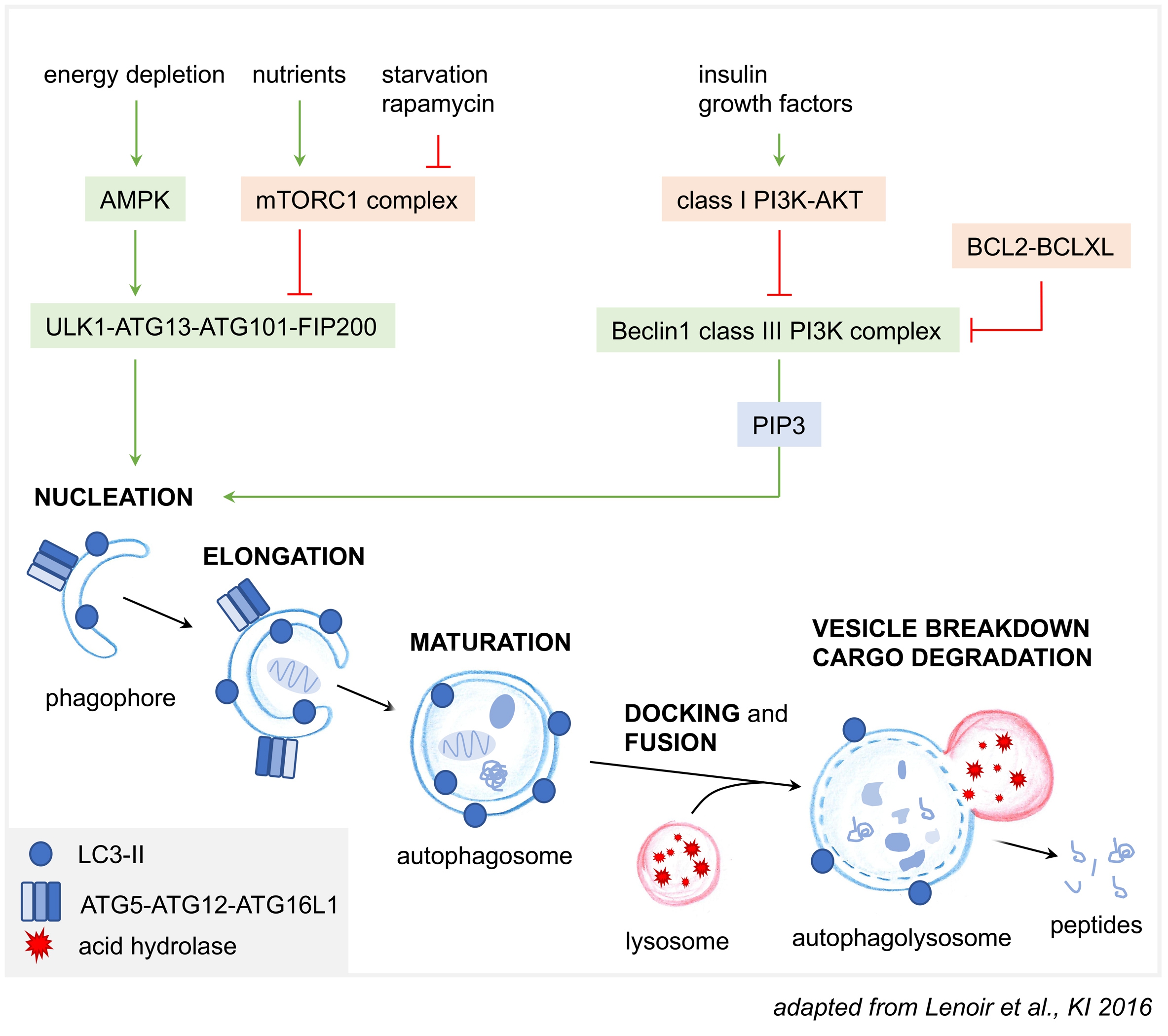
To initiate autophagy at the phagophore assembly site, a range of signaling processes converges on two main kinase protein complexes: the ULK1 (unc51-like autophagy activating kinase 1) protein kinase complex and the Beclin1-PI3KC3-C1 (class III phosphatidylinositol 3-kinase complex I) lipid kinase complex [28]. Multiple levels of (auto)phosphorylation and protein-protein interactions are necessary for activation of the two complexes, with ULK1 and Beclin1 being the respective major sites of regulation. Modification (phosphorylation) of ULK1 and ATG13 results in the recruitment of the activated ULK1 complex (with its components ULK1-ATG13-ATG10-FIP200) to the phagophore assembly site [29], a process also influenced by LC3. ULK1 then transduces pro-autophagic signals by phosphorylating many substrate proteins [29] including itself, subunits of the ULK1 complex and other elements of the core autophagy machinery such as Beclin1 and AMBRA1 (Autophagy and Beclin1 Regulator 1) and hence stimulates the activity of the Beclin1-PI3KC3-C1 complex (with its components Beclin1-VPS34-ATG14L-p150) [30, 31]. The Beclin1-PI3KC3-C1 complex is driven to the phagophore association site by its unique ATG14L subunit. As an essential early event in autophagy initiation, the activated PI3 kinase Vps34 of the Beclin1 complex produces PI3P at the phagophore membrane. The PI3P binding WIPI1-4 proteins and the two ubiquitin like conjugation systems then function downstream and drive phagophore elongation. The first conjugation system comprises LC3-ATG4-ATG7-ATG3 which conjugate phosphatidylethanolamine to free LC3 (LC3-I) in the cytosol. This lipidated LC3 (LC3-II) then redistributes to autophagic vesicles. Therefore, LC3-II is commonly used as a marker for autophagy. The second conjugation system involves the assembly of an ATG12-ATG5-ATG16 complex, which is required for the targeting of LC3 to the double membrane of the expanding phagophore, hence allowing further autophagosome growth, closure and maturation into autolysosomes. LC3-II also mediates the interaction with the cargo-loaded autophagy receptor p62 [32]. In selective autophagy, the cargo itself templates the size and shape of the phagophore. After completed elongation and closure, the outer membrane of the autophagosome fuses with the lysosome to form the autophagolysosome, a process controlled by SNARE proteins [33]. At this stage the inner membrane and all its content are degraded.
Genetic alterations of autophagy and proteasomal genes
In line with their diverging roles and regulation within a cell, genetic alterations of autophagy genes do not comprise cell viability [34], which is in strong contrast to genes encoding proteasome subunits, which are essential [35]. Genetic evidence indicates that both the UPS and autophagy are both implicated in immune modulation. Polymorphisms in autophagy genes are associated with immune diseases involving the kidney, such as polymorphisms in autophagy related 5 gene (ATG5) with systemic lupus erythematosus [36] and the PR domain 1 gene (PRDM1)-ATG5 intergenic region with rheumatoid arthritis [37]. A spectrum of disorders referred to as proteasome-associated auto-inflammatory syndrome (PRAAS) [38, 39] have their origin in mutations of constitutive core particle subunit genes such as PSMA3 (α7) and PSMB4 (β7), in immunoproteasome subunit genes PSMB8 (β5i) and PSMB9 (β1i), and in the general core particle assembly chaperone gene POMP (UMP1) [40]. The common finding in PRAAS is impaired proteasome activity due to CP assembly defects especially in immunoproteasome expressing cells leading to a hyper-inflammatory phenotype with elevated IL-6 and IFN signaling. No renal phenotype in PRAAS has been described so far.
Crosstalk between the two degradation systems
The UPS and autophagy have for a long time been considered to be independent mechanisms of protein degradation. However, the existence of a strong crosstalk has been described between both pathways mainly in the brain [41, 42] but also in podocytes (Fig. 3). Thereby, when the UPS is overwhelmed, inhibited, or when cellular ATP levels are reduced (proteasomal degradation is a highly ATP-consuming process) autophagy steps in [9] to eliminate the accumulating aberrant proteins, which are then sequestered by autophagosomes and delivered to lysosomes for degradation. This cellular reaction is mediated by the unfolded protein response (UPR) through activation of the transcription factor ATF4 [43], by activation of the IRE1-JNK pathway that releases the inhibitory break of Bcl-2 on Beclin1 through phosphorylation [44], and by p53 which accumulates upon proteasomal impairment and thereafter activates AMPK [45] to induce autophagy. The cellular sensors for impaired proteasomal degradation are mitochondria and ER. Thereby, the accumulation of unfolded or damaged proteins alters the mitochondrial proteome, leading to a burst of mitochondrial reactive oxygen species, which activate AMPK to induce autophagy [46]. Additionally, reduced levels of ATP result in enhanced levels of AMP which in turn activate AMPK and autophagy [47]. It has, however, also been reported that continuous impairment of the proteasome can result in an impairment of autophagy [48]. Whether the UPS is activated to compensate for impaired autophagy is debated. The general consensus is that autophagosomal substrates are too large to be channeled through the barrel of the proteasome [49], and that accumulation of p62 and its sequestration to ubiquitinated proteins delays their delivery to proteasomes [50], thereby playing a negative factor in UPS activity [51]. Despite this general consensus, experimental data in podocytes suggest that the UPS is activated in the early course of autophagy impairment (due to genetic ATG5- deficiency) as a compensatory attempt [52]. Only with age does proteasomal impairment add up to the impaired autophagy resulting in proteinuria [52]. Further challenging this general consensus are the findings that proteasomes can indeed clear insoluble protein aggregates, a process mediated by the ubiquitin receptor Ubiquillin 2 in conjunction with the chaperones HSP70-HSP110 i.e. in the nucleus, where autophagy is not present for the removal of protein aggregates [53]. Further, the 20S proteasome is indeed capable of degrading LC3, after detaching from its 19S regulatory particles [54].
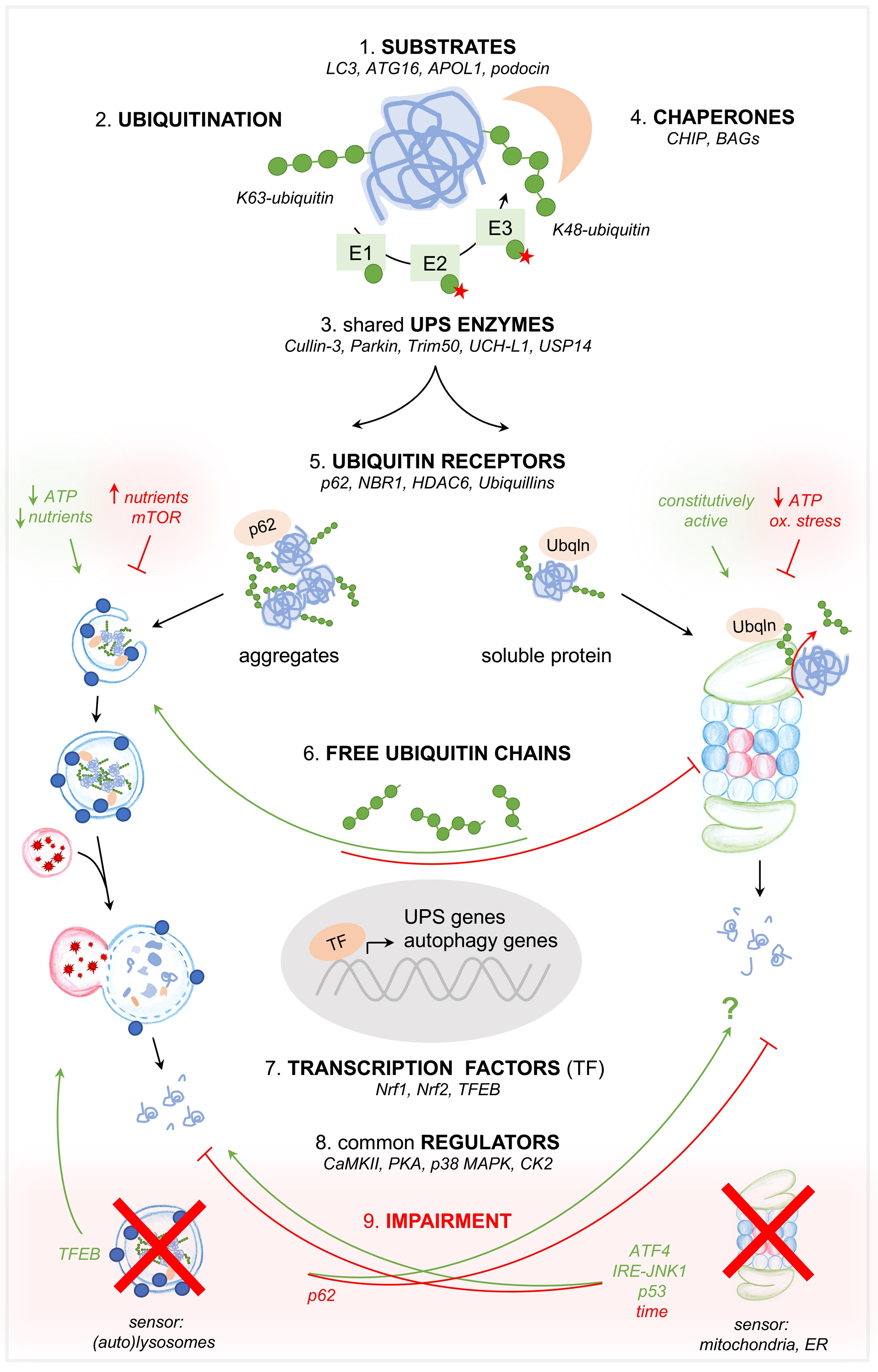
Besides this compensatory crosstalk in the setting of UPS or ALP impairment, both systems are intertwined at 8 further different levels: 1) by common substrates, mechanistically 2) by substrate ubiquitination, 3) by common (de)ubiquitinating enzymes, 4) by chaperones, 5) by ubiquitin receptors, 6) by common transcription factors, and 7) by common regulating kinases. Besides regulating kinases, the effectiveness of both degradation systems is modulated 8) by free ubiquitin chains, which on the one hand impair (clog) the proteasome and thus decrease proteasomal degradation [55], and on the other hand enhance the aggregation of substrates, making them more amenable to autophagy [56, 57].
The intertwining between the UPS and ALP orchestrates the function of the two most basic cellular degradation systems and is therefore relevant to podocyte biology even though not many investigations exist in this specific cell type. Known podocyte-specific proteins that represent common substrates to the UPS and ALP are α-actinin-4, podocin, and nephrin as detailed in paragraph “The UPS and autophagy in the regulation of podocyte identity”. Interestingly, also the Parkinson’s disease associated α-synuclein [58, 59], and the amyloid precursor protein APP, which is cleaved in Alzheimer disease to CTFβ and Aβ [60-62] are common UPS and ALP substrates, which aggregate and accumulate in neurodegenerative diseases. Both substrates are also expressed in the kidney and in podocytes, however their involvement in renal disease is not described to date. From the autophagy perspective, the crosstalk between both degradation systems has been described at the level of the second ubiquitin-like ATG12-ATG5-ATG16 complex required for the lipidation of LC3 and its recruitment to the phagophore or isolation membrane. Thereby, ATG16 binds ubiquitin, is a substrate of the proteasome, and modulates proteasomal activity. Further, ATG16 determines the neddylation of the E3 ligase Cullin-3 (Cul3), which mediates ubiquitination and proteasomal degradation of p62 [63]. A further prominent autophagy-regulating substrate for ubiquitination is the ULK1 (and PI3KC3-C1), which following activation, can be ubiquitinated by the Cul3-KLHL20 ligase complex and degraded [64], thereby switching off the autophagy-initiating signal.
Ubiquitin, a well-known signal for the degradation of polypeptides in the proteasome, also plays an important role in the recognition of cargoes destined for degradation by autophagy [65]. For a long time, the kind of ubiquitin linkage was thought to decide whether a substrate is degraded by the UPS or by autophagy. However, the initial categorization that K48-linked polyubiquitin chains direct proteins to the proteasome and K63-linked polyubiquitin chains direct proteins to autophagy [41] has been questioned, as K48 and K63-polyubiquitin chains are both recognized by autophagy receptors [66, 67] and both bind with comparable affinities to purified proteasomes [68]. Newest studies have elucidated that after substrate ubiquitination, competing ubiquitin receptors harboring either proteasomeor LC3-binding modules determine, which degradative pathway is chosen. Proteasome pathway receptors bind ubiquitin moieties more efficiently, but autophagy receptors gain the upper hand following substrate aggregation and receptor bundling [69]. This enables a triage process, which is thought to depend on the monomeric or oligomeric physical states of these receptors rather than on specific ubiquitin moieties on the substrate [69, 70].
Chaperone proteins such as CHIP (c-terminus of Hsp-70-interacting protein) and BAG (BCL-2-associated anthanogenes) 1 and 3 contain domains, which can direct substrates either to degradation by the proteasome or the autophagosome. Thereby the tetratricopeptide repeat domain of CHIP directs substrates for degradation by the UPS and the U-box domain by the lysosome [71]. BAGs interact with CHIP to channel protein degradation by the UPS [72].
Generally, the autophagosomal degradation of protein aggregates (aggresomes), mitochondria (mitophagy), bacteria (xenophagy), peroxisomes (pexophagy), zymogen (zymophagy), proteasomes (proteaphagy), midbodies, and nucleic acids are considered to represent ubiquitin-dependent mechanisms of autophagy (reviewed in [65]), which are orchestrated by a plethora of so-called ubiquitin receptors. The best-known ubiquitin receptors, which act as bridging molecules to link ubiquitin to autophagy, are p62 [73, 74], neighbor of BRCA1 gene 1 (NBR1), and histone deacetylase 6 (HDAC6) [75, 76]. These proteins contain ubiquitin associated (UBA) domains specialized for binding to ubiquitin and an additional domain dedicated to linkage to autophagosomes, such as the LC3 interacting region (LIR) that facilitates adaptor protein binding to LC3 [77]. Examples for ubiquitin receptors that mainly target ubiquitinated proteins to the proteasome are Rpn13 [78], Rpn10 [79], Rpn11 [80], or Ubiquillin 2 for the clearance of protein aggregates by the proteasome [53]. Interestingly, Rpn11, Ubiquillin 2 and p62 have been shown to also mediate substrate degradation by both autophagy and the UPS [57, 81, 82]. Thereby p62 was the first autophagy receptor described to be essential for clearance of ubiquitinated substrates in mammals, especially in the setting of UPS inhibition [83, 84]. In situations of ubiquitin stress, that is in situations of ubiquitin overexpression, heat shock or prolonged proteasomal inhibition, p62 undergoes E2-dependent ubiquitination liberating its ability to recognize polyubiquitinated cargoes for selective autophagy [85]. P62 is upregulated by the transcription factor Nrf2 [86] and binds ubiquitin via its UBA domain [87]. Following oligomerization, p62 targets ubiquitinated substrates to autophagy [83], a process (not necessarily [88]) mediated by its association with LC3 [89] and with GABARAP subfamilies on the autophagosome [73, 83, 89]. Additionally, p62 can also target ubiquitinated proteins to the 19S RP of the proteasome for proteasomal degradation [81]. The ubiquitin receptor Rpn11 on the other hand, normally involved with the deubiquitination of the vast majority of proteins that enter the 20S CP, has been shown to target aggresomes in a coordinate action with unanchored K63-chains to the microtubule-organizing center for degradation by autophagy [12, 56]. Unanchored K63-ubiquitin chains are further important for the fusion of autophagosomes with lysosomes [56, 57].
The UPS and autophagy share enzymes involved in the (de)ubiquitination process. As such, the E3 ligase parkin targets proteasomal substrates with K48-polyubiquitin chains [90] and lysosomal substrates with K27-polyubiquitin chains [91]. Parkin senses the functional state of mitochondria and directs damaged mitochondria for disposal via mitophagy by building ubiquitin chains on the mitochondrial outer membrane proteins, where they act to recruit autophagy receptors such as NDP52 and optineurin [92, 93]. Both systems share the E3 ligase TRIM50, which is necessary for the clearance of aggresomes [94]. Besides common E3 enzymes both systems share deubiquitinating enzymes, such as UCH-L1 and USP14. UCH-L1 is thought to maintain the pool of monoubiquitin required for ubiquitination procedures [95], and is also involved in the lysosomal degradation of BACE1, the β-secretase cleaving APP to CTFβ [96, 97]. USP14 associates with the proteasome 19S RP and deubiquitinates substrates prior to their proteasomal degradation, thereby regulating proteasomal activity [98]. Upon proteasome inhibition, association of USP14 with the 19S RP is disrupted resulting in an enhanced interaction of USP14 with the autophagy protein GABARAP and enhanced autophagy [99]. USP14 also regulates autophagy by suppressing the K63 ubiquitination of Beclin1 [100].
Finally, posttranslational modifications achieved by kinases have been demonstrated to represent an additional level of regulation at which the crosstalk between the UPS and autophagy is orchestrated. The most prominent kinases, which influence both proteasomal and autophagy activity in a concordant or inverse direction, are the serine/threonine kinase casein kinase 2 (CK2), the Ca2+/calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA) and p38 MAPK [42]. Thereby, CK2 phosphorylates the α7 subunit of the proteasome and thereby regulates proteasome assembly and opening [101, 102]. CK2 also modulates autophagy by phosphorylation of ATG32, a process essential for mitophagy [103]. CaMKII enhances proteasomal activity by phosphorylation of the proteasomal rpt6 subunit of the 19S RP [104, 105]. CaMKII activates autophagy by direct phosphorylation of Beclin1 to promote K63-linked ubiquitination of Beclin1 [106]. Activation of PKA through increased cAMP levels enhances phosphorylation of Rpn6 and Rpt6 subunits of the 19S RP. These posttranslational modifications are required for the correct assembly of the 26S proteasome by interaction of rpt6 with the α2 subunit of the 20S core particle [107] and for an enhanced proteasomal activity [108]. Autophagy is suppressed by elevated levels of PKA activity at the ATG1/ATG13 kinase complex [109], and a feedback loop of autophagy to PKA regulation is suggested [110].
Protein degradation systems in podocyte physiology
The relative contribution of autophagy and of the proteasome system in podocyte proteostasis is unclear as the mere histological detection of UPS and ALP players does not assess the overall activity state of the respective degradative systems. The proteasome abundance, proteasome degradative capacity and selectivity are not fixed within a cell. They are precisely regulated by transcription factors, by proteasomal assembly chaperones and by posttranslational modifications to adjust to physiological needs. It is presumed that cells contain an excess of unengaged proteasomes, which can be assembled and mobilized when needed [111]. Some regulators affect proteasomal activity by associating with it, others modify the proteasome post-translationally, and still others promote the transcription of proteasomal genes [112]. Triggers that lead to changes in proteasomal abundance and subtypes are multiple and include: 1) cytokines, with IFNγ being the most potent one, 2) oxidative stress 3) ER stress activating ER associated degradation system, and 4) enhanced protein synthesis. In contrast to the proteasome, the cellular levels of autophagy vary considerably depending on the cell type and on the nutrient supply, with energy deficiency and starvation being major inducers of autophagy aiming at restoring intracellular ATP and nutrient supply such as amino acids for protein synthesis [113]. It is generally accepted that (if nutrients are abundant), a cell can survive without autophagy, as the amino acid supply is constitutively provided by the actions of the UPS whose protein turnover is constantly high. Autophagy is regulated by 4 main growth-signaling pathways converging either at the ULK1 complex or at the Beclin1-PI3KC3-C1 complex: 1) AMP-activated protein kinase (AMPK) initiates autophagy in the setting of energy depletion, 2) mammalian target of rapamycin (mTOR) complex 1 (mTORC1) inhibits autophagy in the setting of high nutrient, amino acid glucose supply, 3) the anti-apoptotic factor B-Cell lymphoma 2 (Bcl-2) inhibits autophagy, and 4) class I PI3K-AKT inhibits autophagy following stimulation with insulin and/or growth factors. Besides energy and nutrient supply, laboratory and clinical evidence highlight ER stress and hypoxia as strong modulators of autophagy, excellently reviewed in [114-116].
Cellular levels of UPS and autophagy activity in podocytes
Podocytes are terminally differentiated cells and therefore their capacity to regenerate is limited [117]. Similarly to neurons, they require efficient cellular mechanisms to remove flawed proteins that accumulate over their lifetime, cellular debris and damaged organelles. Autophagy seems to be one of these cleaning mechanisms that ensure podocyte maintenance, especially with age. In line, podocytes are sensible to autophagosomal inhibition [118]. Experiments with GFP-LC3 reporter mice suggest that podocytes have high levels of basal autophagy [51, 119] in comparison to other intraglomerular cell types. Nonetheless, levels of lysosomes and activity of lysosomal acid hydrolases required for the degradation of autophagosomal content after autophagolysosome formation have repeatedly been shown to be higher in glomerular endothelial and mesangial cells than in podocytes in unstressed conditions in multiple pioneer studies [120-122]. Despite this conundrum, prevention of autophagosome formation through podocyte-specific knockout of ATG5 results in proteinuria, loss of podocytes and age-related glomerulosclerosis in mice aged older than 7 months. Interestingly, prior to the development of these symptoms, younger podocyte-specific ATG5-deficient mice upregulate the UPS in podocytes, compensating for the loss of autophagy. With 7 months however, the UPS becomes impaired, as proteasomal activity decreases and polyubiquitinated proteins accumulate, a finding coinciding with the manifestation of podocyte injury in podocyte-specific ATG5-deficient mice [52] demonstrating that both degradative systems are imperative for podocyte maintenance. To which extent is however unclear. In some instances of ALP impairment, podocytes exhibit severe injury and loss, while in others, podocyte injury is mild to absent possibly due to a compensatory upregulation of proteostatic stress reducing pathways. For instance, mice with a podocyte-specific deficiency of Vps34, which in yeast is essential for the sorting of hydrolases to yeast vacuoles [123] and in mammals (as mVps34 (class III PI3K)) has been implicated in the regulation of autophagy [124], develop severe glomerulosclerosis with enlarged vacuoles and increased autophagosomes in podocytes and die at 9 weeks of age [125]. This phenotype appears to be the result of a direct lysosomal impairment rather than of a primary defect in autophagy. On the other hand, mice with the lysosomal storage disease mucolipidosis type II or III do not exert overt podocyte injury. This lysosomal storage disease is due to mutations of the Golgi resident GlcNAc-1-phosphotransferase. Mutations in GNPTAB result in inactive GlcNAc-1-phosphotransferase cause the severe MLII disease, whereas a few mutations in GNPTAB and mutations in GNPTG, which are accompanied by a residual GlcNAc-1-phosphotransferase activity, cause the less progressive diseases MLIII alpha/beta or MLIII gamma, respectively [126]. Consequently, in cells from patients with MLII and MLIII, the generation of M6P targeting signals on lysosomal enzymes is impaired and leads to their missorting and hypersecretion into the extracellular compartment [127]. The subsequent intracellular deficiencies of multiple lysosomal enzymes result in the accumulation of various non-degradable macromolecules in dysfunctional lysosomes in all glomerular cell types [128]. Interestingly, as in ATG5 deficiency, the glomerular UPS compensates for cellular homeostasis in MLIII mice, whereas in MLII with the severe lysosomal enzyme missorting protein synthesis through the integrated stress response was decreased to alleviate for the disturbance in proteostasis [128].
In cultured murine and human podocytes, proteomic studies demonstrate a strong proteolytic shift from a predominant proteasomal activity to a lysosomal activity in the course of podocyte differentiation [129, 130], which coincides with a globally increased stability of mitochondrial, cytoskeletal, and membrane proteins in differentiated podocytes [130]. How far this finding can be transferred to the physiologic in vivo situation still needs to be determined, as possible skewers such as culture conditions could explain the proteolytic system shift from undifferentiated to differentiated podocytes. Recent data rom our lab suggest that podocytes have an exceptional high dependence on a functioning proteasomal system in vivo, already at young age. Thereby, a new technique enabling a reporter-free bulk separation of glomerular cell-types from wildtype mice for protein biochemical investigations shows that podocytes express higher levels of proteasomal proteins in comparison to mesangial and glomerular endothelial cells [131] in 8 to 10-weekold mice. The strong podocyte dependence on the proteasome for regular proteostasis is further stressed by the finding that podocyte-specific deficiency of rpt3, a subunit of the 19S RP of the proteasome leads to early onset podocyte injury (albuminuria at 4 weeks of age) and loss, with atrophic kidneys at 14 weeks of age [132]. More investigations are required to dissect the individual contribution of the UPS and ALP for podocyte proteostasis.
However, sub summation of the current literature lets us suggest that podocytes exhibit a basal dependence on the proteasome system, which steps in to alleviate proteostatic stress in the setting of ALP impairment, whereas autophagy appears to be especially relevant for podocyte homeostasis in aging and in the setting of podocyte stress.
The UPS and autophagy in the regulation of podocyte identity
Accumulating evidence suggests that homeostatic levels of podocyte-specific proteins are dependent on the UPS [18], in some cases in tight interplay with the endocytic / autophagosomal system. The development and maintenance of podocyte identity is carried out by the major podocyte transcription factor WT1, which is responsible for regulation of an extensive transcriptional network of podocyte-specific proteins [133]. The UPS is involved in WT1 ubiquitination and degradation in a β-catenin dependent manner [134], thereby strongly affecting podocyte identity. Proteomic analyses have further identified endogenous ubiquitination sites on podocyte-specific proteins such as Neph1 (also known as KIRREL1), α-actinin 4, and synaptopodin suggesting an involvement of the UPS in their levels and distribution [135]. In line, levels of synaptopodin, α-actinin 4, and nestin can be stabilized by proteasomal inhibition in parietal epithelial cells (which share common lineage to podocytes) [136] or in podocytes in the setting of injury [9]. Induction of autophagy was shown to regulate podocalyxin levels in parietal epithelial cells [136]. Further underscoring an involvement of the proteasome in the maintenance of homeostatic levels of podocyte essential proteins, the UPS was shown to be necessary for the ubiquitination and removal of mutant α-actinin-4 [137] and podocin [138]. However, again an intact interplay with the ALP was shown to be necessary. In case of mutant α-actinin-4, autophagy is increased in glomerular epithelial cell cultures, presumably to relieve ER stress and remove α-actinin-4 aggregates [139] secondary to proteasome impairment [137]. Podocin and nephrin are both ubiquitinated after growth factor stimulation in CD2AP negative podocytes, a process dependent on CIN85/RUKL, a mediator of ubiquitin-mediated endocytosis. This process is thought to destabilize the slit membrane in the absence of CD2AP resulting in endocytosis of nephrin [140]. Nephrin endocytosis is widely accepted as a means for nephrin regulation at the slit membrane [141]. To enhance nephrin stability at the slit membrane, modification of nephrin by the ubiquitin-like protein SUMO at the intracellular tail was shown to be essential. SUMO binds at lysine residues to block ubiquitination of the same site leading to stabilization of the target protein [142]. Podocin on the other hand is polyubiquitinated by the E3 ligase Ubr4 [135]. Interestingly, both K48- and K63-polyubiquitination of podocin have been observed resulting in unfolding and proteasomal as well as lysosomal degradation of podocin [135]. Synaptopodin regulates the podocyte actin cytoskeletal dynamics by associating with two E3 ligases. First, synaptopodin regulates stress fiber formation by interfering with the binding of the E3 ligase Smurf1 to the actin-cytoskeleton regulator RhoA, thereby preventing its ubiquitination and degradation [143]. Second, synaptopodin also competes with the E3 ligase Cbl for binding to the adaptor protein Nck1 located at the slit membrane, which is involved in the phosphorylation and endocytosis of nephrin. The interaction of synaptopodin with Nck1 decreases the ubiquitination and proteasomal degradation of Nck1 [144]. These findings underscore a primary function of the UPS in maintaining a podocyte identity by regulating homeostatic levels of podocyte-specific proteins, in some cases in tight interplay with the endocytic / autophagosomal system.
Proteolytic systems in podocyte stress response
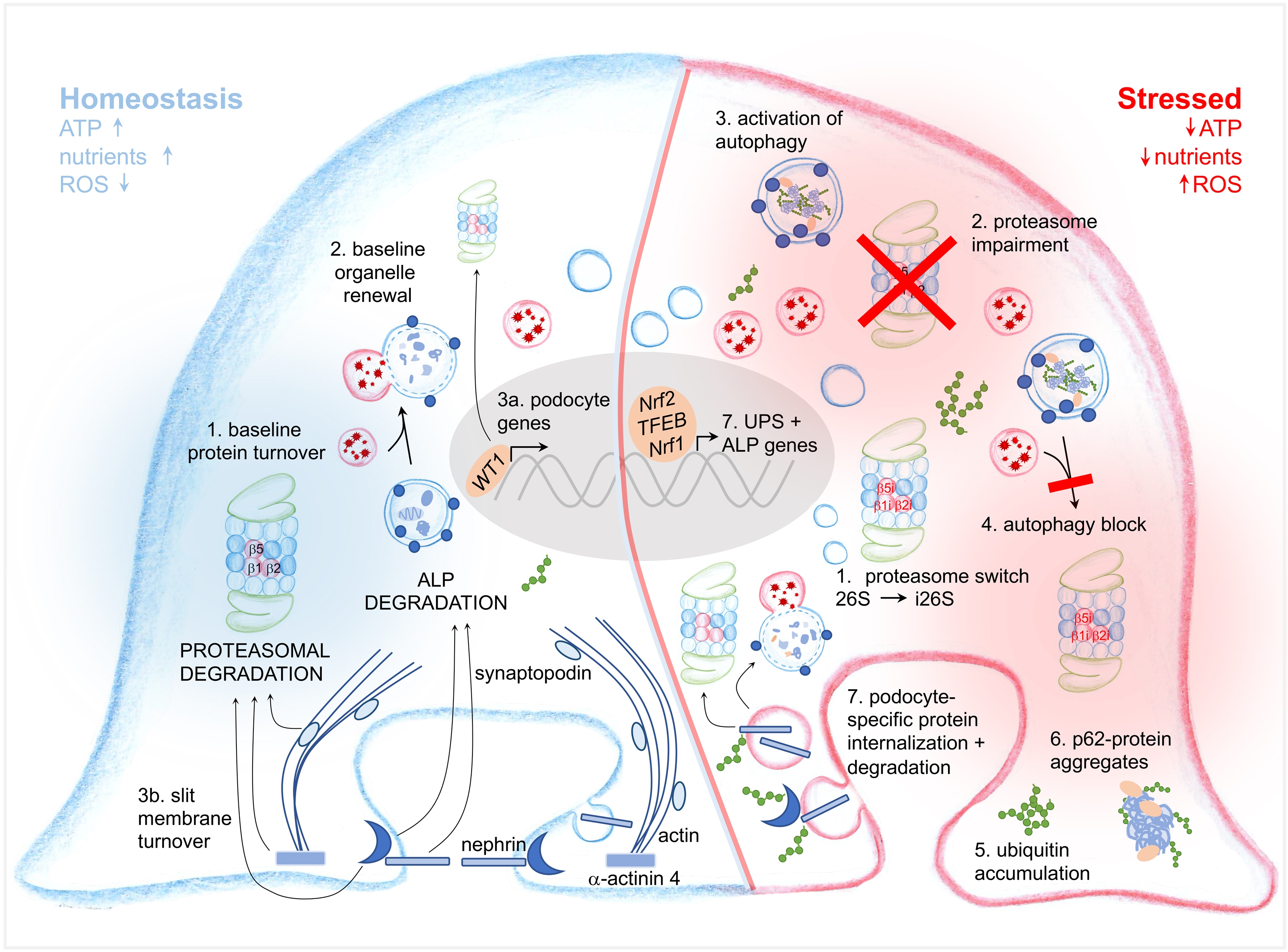
As differentiated post-mitotic cells, podocytes strongly depend on their ability to react to different situations of cellular stress, e.g., proteotoxic or oxidative stress. Findings implicating an involvement of proteostatic systems in podocyte injury have been reported for both the UPS and the ALP as reviewed elsewhere [9, 18, 114]. Depending on the species, type and stage of kidney disease, reported findings may be contradictory and experimental evidence for some of the observations are still scarce/missing, partially due to the complex nature of the degradative systems. As a solidly validated general theme (Fig. 4), autophagy is induced in most situations of podocyte injury, highlighting its importance for stress-related balance of proteostasis. Impairment of autophagy has also been described in podocyte injury, there mostly as a long-term sequel rather than as a primary event. Research on proteasomal function in podocyte injury is scarce to absent. First publications, however, suggest an initial reaction of the UPS to limit podocyte injury [9]. Early UPS reactions comprise an enhancement of proteasomal activity partly by upregulating and changing proteasome subtypes from the standard 26S proteasome to the i26S immunoproteasome and an enhanced expression of ubiquitin and UPS enzymes [9, 145]. Proteasomal function appears to be rapidly impaired in stressed podocytes by unidentified factors. This is reflected by the accumulation of oxidative-modified (podocyte-specific) proteins and by K48-polyubiquitin, and by decreased proteasomal activity. Whether induction of autophagy in stressed podocytes is an independent mechanism to the UPS to limit podocyte injury, or whether it mostly occurs in crosstalk with the UPS to compensate for proteasomal impairment is not known and certainly strongly dependent of the type of inflicted podocyte stress. One aspect that is neglected in many studies is the heterogeneity of glomerular cell types. Often, protein or mRNA levels are assessed on glomerular or even renal level, making conclusions towards one specific cell type and the actual activity of the system hard to draw. In the following sections, the evidence already available for some common glomerulopathies are summarized and new developments highlighted that might facilitate new discoveries in the near future.
Ageing
Accumulation of damaged organelles such as mitochondria as well as protein aggregates is the common theme of ageing cells, a process from which podocytes are not exempt. Recent comparative measurements of the transcriptomic signature of young and aged murine podocytes exhibit differential regulation of cellular pathways, which affect or are involved in the regulation of autophagy and the proteasome, such as the PI3K-mTOR pathway and the p53 pathway [146]. As a main theme, however, this study revealed an increase of apoptosis and senescence related transcripts in aged podocytes [146]. Nonetheless, endocytosis transcripts were enhanced in aged podocytes [146], possibly resulting in enhanced autophagy. The downregulation of the electron transport chain transcripts and of the MAP kinase activity transcripts [146] could relate to impaired proteasome function. In line, experimental data support the importance of autophagy as an essential cleaning mechanism allowing podocyte maintenance during ageing. Atg5 knockout podocytes show typical agerelated alterations, namely lipofuscin accumulation, an increase in oxidized proteins, and ubiquitin and p62 positive protein aggregates [52]. The contribution of the UPS in ageing podocytes is completely unclear. Does proteasomal impairment occur, similarly to ageing neurons? In old mice podocytes show higher levels of UPS players such as ubiquitin and UCH-L1 in comparison to younger mice, which might be indicative of both, proteasomal impairment or UPS activation. Since loss of UCH-L1 in podocytes results in an enhanced accumulation of oxidative-modified proteins and K48-polyubiquitinated proteins in 22-24 months old mice (the equivalent of 70-year-old humans), at least the upregulation of this deubiquitinating enzyme could be considered as a protective mechanism to age-related proteostatic disturbances in podocytes [145].
Diabetic nephropathy
Diabetic nephropathy (DN) is a renal complication occurring with both type 1 and type 2 diabetes that may result in end-stage kidney disease. Classical clinical symptoms like proteinuria and elevated serum creatinine levels can be observed. While the observed effects on renal function match those of other glomerulopathies, the molecular mechanisms linking high blood glucose levels to renal failure are incompletely understood. However, observations implicating both the ALP and the UPS in DN have been described and reviewed [147, 148].
High glucose levels markedly increase the autophagic flux in glomeruli of diabetic mice and in cultured podocytes (both primary and immortalized). An impairment of this autophagic response via podocyte-specific knockout of Atg5 leads to higher apoptosis rates under high glucose conditions in cultured podocytes and exacerbates clinical parameters in a mouse model of DN. Interestingly, this initial upregulation of autophagy was reversed upon long-term high glucose conditions, resulting in lower levels of autophagy at the time of glomerular lesions forming. Podocyte-specific loss of Atg5 in mice resulted in higher apoptosis rate, increased thickening of the GBM and substantiated glomerular morphological changes under high glucose conditions [149]. Further studies confirmed these findings in kidney biopsies of DN patients and gathered evidence that serum factors are involved in the downregulation of podocyte autophagy under DN conditions. The accumulation of damaged lysosomes possibly indicate impairment of lysophagy as one crucial process. Importantly, downregulation of autophagic flux was only observed in the presence of proteinuria and not in other disease stages [150]. These findings were further substantiated by observations that induction of autophagy by resveratrol and other pharmacologic agents protects podocytes from high glucose-induced apoptosis [151, 152]. In a complex interplay between glomerular endothelial cell and podocyte damage, podocyte autophagy was shown to be protective against damage to glomerular endothelial cells. As such, inducible deletion of Atg5 in podocytes rendered podocytes more susceptible to structural damage when they were located adjacent to injured glomerular endothelial cells in a model of high fat diet [153].
Upregulation of UPS activity under high glucose conditions was also reported in numerous models of DN. However, in contrast to the autophagic degradation pathway, inhibition of the UPS has been shown to ameliorate the disease course. In a streptozotocininduced model of DN in rats, proteasomal inhibition with MG132 improved clinical parameters and relieved oxidative stress in the kidney cortex [154]. Following up on this finding, in the OVE26 mouse model of type I diabetes, non-toxic doses of MG132 resulted in lowered proteasomal degradation of the transcription factor Nrf2, which in turn increased the expression of several enzymes with anti-oxidative activity like SOD2 and HO-1 [155]. Mediated by NF-κB, MG132 repressed Akt-mediated inflammation and thereby protected mice against renal dysfunction in DN [156]. However, these effects were only assessed in whole kidneys and evidence for specific glomerular cell types is still lacking. Since complex interactions between glomerular cells have been observed in other contexts [1, 157], these questions need to be addressed in a glomerular cell-specific manner in future studies.
Ubiquitin independent UPS-mediated degradation of the SNARE proteins syntaxin 17 (STX17) and SNAP29 negatively regulates autophagic flux by preventing the necessary vesicle fusion between autophagosomes and lysosomes [158]. This effect might add to the compensatory upregulation of autophagy under proteasomal impairment and could add to the reasons why inhibition of the proteasome is beneficial in DN models. The decreased degradation of the transcription factor Nrf2 upon proteasome inhibition results in its nuclear translocation, increasing the transcription of, among other genes, p62 and HO-1, which could further result in increased autophagic flux [155, 159]. While several such hints exist towards an essential role for UPS-ALP crosstalk in podocytes in the development of DN, concrete evidence is still lacking and will have to be supplied by future studies. Observations that degradation of accumulating proteins via autophagy is triggered via aggregation and receptor bundling could indicate that autophagy simply takes over when the UPS has failed to discard (damaged) proteins [69]. The open conformation of K63-conjugated ubiquitin chains could facilitate aggregation and receptor bundling more easily than the compact chains formed by K48-conjugated polyubiquitin [160, 161]. Mitochondria have been described to be involved in the sensing of UPS impairment and in AMPK-mediated upregulation of autophagy [162]. Together with findings that in podocytes, autophagy is mainly regulated through the AMPULK1 axis and not via mTOR signalling, this indicates a possible crosstalk mechanism in podocytes [163].
Focal segmental glomerulosclerosis
Focal segmental glomerulosclerosis (FSGS) is a generic term for a histological injury pattern defined by segmental glomerular scarring that affects some but not all glomeruli with a wide range of etiological interpretations, as FSGS describes both a disease characterized by primary podocyte injury (primary FSGS), and a lesion that occurs secondarily in any type of chronic kidney disease (secondary FSGS) [164]. Up to 50 causative mutations to podocyte proteins have been identified as causes to primary FSGS [165], one of which is the lysosomal integral membrane protein (LIMP) 2 [166]. In micro-dissected human glomeruli, UPS and ALP transcript upregulation was only present in FSGS and not in minimal change disease (MCD) [9], a histological condition presenting with to FSGS comparable morphological podocyte alterations, however importantly without glomerular sclerosis. Further underlining a differential pathogenic involvement of the protein degradation systems in FSGS and MCD was the observation that the presence of autophagosomes in podocytes differentiated patients from these two groups in renal biopsies. Autophagosomes were detected (via electron microscopy) in significantly more podocytes from MCD patients than in those from FSGS patients. Analysis of LC3 and Beclin1 protein levels substantiated these findings. Repeat renal biopsies of MCD patients further hint towards a protective role of autophagy. While in patients that progressed from MCD to FSGS, autophagosome numbers decreased significantly, this was not the case in patients with persistent MCD [167].
FSGS is mimicked in rodents by inducing selective podocyte injury either by administration of chemical substances such as puromycin or by genetically inducing podocyte loss [168]. These animal models are the basis of investigations showing an upregulation of ALP and UPS players [9] and of suggesting a beneficial effect for autophagy modulation in FSGS. Thereby, inhibition of autophagic flux with 3-Methyladenine (3-MA) in a rat model of puromycin amino nucleoside (PAN)-induced podocyte damage accentuated morphological changes and proteinuria. Conversely, induction (and maintenance) of high levels of autophagy using rapamycin prevented podocyte foot process effacement and improved proteinuria [167]. Studies in cultured human podocytes revealed an initial upregulation of autophagy upon PAN-induced podocyte damage. Interestingly, as already observed in settings of DN, autophagy declined again over time also in this model of podocytopathy. Inhibition of autophagy via knockdown of Beclin1 lead to decreased levels of podocyte marker proteins and increased podocyte apoptosis, which was also the case when autophagy was inhibited with 3-MA or chloroquine [167]. The role of the UPS in FSGS is mostly related to those FSGS forms induced by mutations of podocyte immanent proteins [165] such as α-actinin-4 and podocin described above, as their removal primarily depends on the proteasome system. Of note, proteasomal inhibition can partially redistribute the FSGS-associated mutant podocin proteins to the plasma membrane in cultured podocytes [138].
The intricate interplay of the UPS and ALP in podocyte pathophysiology is nicely observed in humans of recent African ancestry, which exhibit an increased susceptibility to developing FSGS due to implicated variants of apolipoprotein L1 (APOL1) [169]. Experimental expression of the APOL1 risk alleles APOL1-G1 and -G2 in a podocyte-specific manner is causal for podocyte foot process effacement, proteinuria, and glomerulosclerosis [170]. As a secreted protein, APOL1 can induce lysosomal membrane depolarization resulting in a block of autophagic flux [171] due to impaired autophagosomal – lysosomal fusion ultimately leading to inflammatory-mediated podocyte death and glomerular scarring [170]. On the other hand, sequence variation at the UBD gene locus (also known as FAT10, which encodes an IFNγ-inducible ubiquitin-like protein [172]) was identified as a genetic modifier of the risk of FSGS development in individuals with a high risk APOL1 genotype [173]. Thereby, UBD modification of APOL1-G1 and APOL1-G2 was shown to decrease their levels by proteasomal degradation and thereby mitigate their cytotoxic effects. As African ancestry at the UBD locus is associated with lower levels of UBD transcripts, the impaired proteasomal removal of APOL1-G1 and -G2 in podocytes most likely contributes to the higher risk of FSGS development [173].
Membranous nephropathy
Membranous nephropathy (MN) is an autoimmune disease caused by autoantibodies directed against podocyte foot process proteins, such as phospholipase A2 receptor 1 (PLA2R1) [174] and thrombospondin type-1 domain-containing protein 7A (THSD7A) [175]. Morphologically, MN is characterized by subepithelial immune deposits at the glomerular basement membrane (GBM), resulting in an overall increased GBM thickness. Further, subepithelial deposition of the autoantigen, IgG (mostly IgG4), and complement factors are morphologic characteristics of MN. The mechanisms that initiate podocyte injury after autoantibody binding to foot process antigens remain obscure. Sublytic complement induced injury [176], and mechanical or antigen-related effects are discussed [177]. Downstream of these initiating events, protein degradation pathways are induced in human and rodent MN [9, 178]. Typically, podocytes exhibit extensive hypertrophy and vacuolization, which might partly be attributed to a strong induction of the ALP in MN, as mirrored by transcriptional studies in micro-dissected human glomeruli [9] and by the prominent induction of Limp2-expressing lysosomes [179], and by an enhanced expression of the lysosomal acid hydrolase cathepsin D [180]. Interestingly, lysosomal acid hydrolases are suggested to be involved in the slow degradation of the deposited IgG in MN [181]. Whether the upregulation of the ALP in MN represents a podocyte injury promoting or an attenuating event is not clear. Deficiency of lysosomal Limp2 in mice exacerbates podocyte injury and disease in the antipodocyte nephritis model [9] (a murine model of MN [182]), suggesting that lysosomal induction might represent a disease attenuating response. In line, autophagy has been implicated in the relief of ER stress in podocytes in a rat model of MN, the passive Heymann nephritis, and in cultured murine podocytes [183]. As an indication of induced but defective autophagy in MN related podocyte injury, renal biopsies of MN patients exhibit accumulation of LC3 and p62 [184]. Studies in cultured mouse podocytes link complement, especially the C5b-9 membrane attack complex (MAC), to impaired autophagy through permeabilization of the lysosomal membrane, which leads to deacidification of the lysosomal milieu and to low activity of lysosomal degradative proteases. As a direct cause, autophagosomal flux is impaired and autophagosomal vacuoles accumulate [184]. Interpretation of this result is complicated by the fact that the complement system was also shown to induce the UPS activity via activation of c-Jun N terminal kinases (JNKs), in the setting of impaired endoplasmic reticulum associated degradation (ERAD) [185]. The authors speculate that this indicates a higher ubiquitination activity rather than an actual increase in proteasome catalytic activity [185].
Emerging evidence suggests that the UPS plays a major role in the development and progression of MN. Transcripts of UPS components are upregulated in micro-dissected glomeruli of MN patients, and a podocyte accumulation of ubiquitinated proteins is seen in human MN biopsies [9, 178] and rodent MN [186]. In line with the ubiquitin accumulation in podocytes, proteasomal function was observed to become impaired following induction of anti-podocyte nephritis in UBG76V-GFP transgenic mice (reporter mice that visualize proteasome activity by means of GFP accumulation). Of note, time-course analyses depicted an impairment of proteasome function prior to the development of proteinuria and prior to morphologic podocyte injury [9]. Upregulation of the ALP was a late event in anti-podocyte nephritis, suggesting a compensatory effort of the ALP to restore proteostasis in the setting of proteasomal dysfunction [9]. The UPS is involved in the degradation of oxidatively damaged proteins such as α-actinin 4 in the course of passive Heymann nephritis [9], a degradative pathway that may also apply to other disease associated podocyte foot process proteins, such as THSD7A or PLA2R1, which accumulate in glomeruli of MN patients. Interestingly, specific UPS components could be involved in the pathogenesis of MN. Chemical inhibition of the neuronal specific deubiquitinating enzyme UCH-L1, was shown to prevent podocyte ubiquitin accumulation and to ameliorate the clinical course of MN in passive Heymann nephritis, whereas treatment with the proteasomal inhibitor MG132 further enhanced the ubiquitin accumulation and exacerbated the clinical course [186]. UCH-L1 is thought to maintain the pool of free ubiquitin in neurons [95] and is normally not expressed in healthy podocytes, however de novo expressed in podocyte injury, especially in MN [178, 187, 188]. Whether UCH-L1 perpetuates or attenuates podocyte injury in MN is unclear, as its effects on podocyte proteostasis vary depending on the MN model used (mouse or rat) and on the mode of UCH-L1 modulation (chemical inhibition of hydrolase function versus conditional or podocyte-specific knockout). UCH-L1 was shown to regulate TNF-induced necroptosis in cultured murine podocytes [189], to be important for the balance between proteasome subpopulations in the murine kidney [145], and to influence the glomerular filtration rate [190] of constitutive UCH-L1 deficient mice. Highlighting the importance of UCH-L1 for the regulation of glomerular cell proteostasis with ageing is the observation that a constitutive knockout of UCH-L1, as well as a podocyte-specific knockout of UCH-L1 both resulted in the accumulation of oxidatively-modified proteins under basal conditions in glomerular cells of old mice, especially in podocytes [145]. Correlative observations in rodent models of MN associate UCH-L1 with an overall decreased proteasomal activity [191] and with the development of podocyte hypertrophy [191], a feature that might be related to a described toxic-gain-of-function of this enzyme upon post-translational modification especially induced by oxidative stress [192], or with its influence on the degradation of disease relevant proteins such as the cell cycle inhibitor p27Kip1 [192].
Lupus nephritis
Culminating evidences demonstrates the effectiveness of proteasomal inhibitors in targeting B and plasma cells in antibody mediated kidney injury such as ANCA-associated vasculitis, transplant nephropathy and lupus nephritis (LN) [193], and suggest an advantage of subunit specific targeting of the immunoproteasome [194, 195]. However, analyses of the effects proteasomal inhibitors could exert on resident renal cells such as podocytes in the setting of antibody-mediated injury have not been the focus of present studies. On the other hand, direct evidence suggests a protective effect of autophagy in podocyte injury in the MRL (lpr/lpr) mouse model of LN [196]. In this genetic model of LN, autophagosomes were present in podocytes, but not in glomerular endothelial or mesangial cells. Interestingly, autophagy could be induced in podocytes by treatment with IFNα and IgG isolated from LN patients in this study. Patient IgG appeared to have a direct cytotoxic effect on cultured human podocytes, as exposure to patient IgG led to an increased production of reactive oxygen species and podocyte apoptosis. Apoptosis could be prevented via stimulation of autophagy, whereas autophagy inhibition via 3-MA or bafilomycin aggravated podocyte apoptosis. Exposure to IFNα alone did not lead to increased podocyte apoptosis rates but decreased podocin levels in cultured podocytes and resulted in higher albumin leakage in an in vitro albumin flux assay, representing structural podocyte damage [196]. As IFNα has been shown to be secreted from tubular cells in an autocrine manner in LN and to induce the expression of genes involved in protein ubiquitination and an induction the i26S immunoproteasome [197], IFNα could be responsible for activation of proteasomal (especially of immunoproteasomal) degradation in podocytes in the setting of LN, which might be involved in the decrease of podocyte podocin levels.
Conclusion
Podocytes, as terminally differentiated cells, highly depend on intact proteolytic machineries. Sub summation (Table 1) of common knowledge about the UPS and autophagy in other cellular systems with the few investigations performed in podocytes support the notion that basal high levels of UPS activity predominantly maintain podocyte health, whereas autophagy compensates for UPS impairment during ageing and in podocyte injury, especially in situations in which ATP-depletion and oxidative stress are high in podocytes. The fact that both degradation systems are intertwined at multiple steps renders analyses and interpretations of their contribution to podocyte health and disease challenging. Future investigations in this fascinating field will open new avenues to therapeutically modulate podocyte injury.
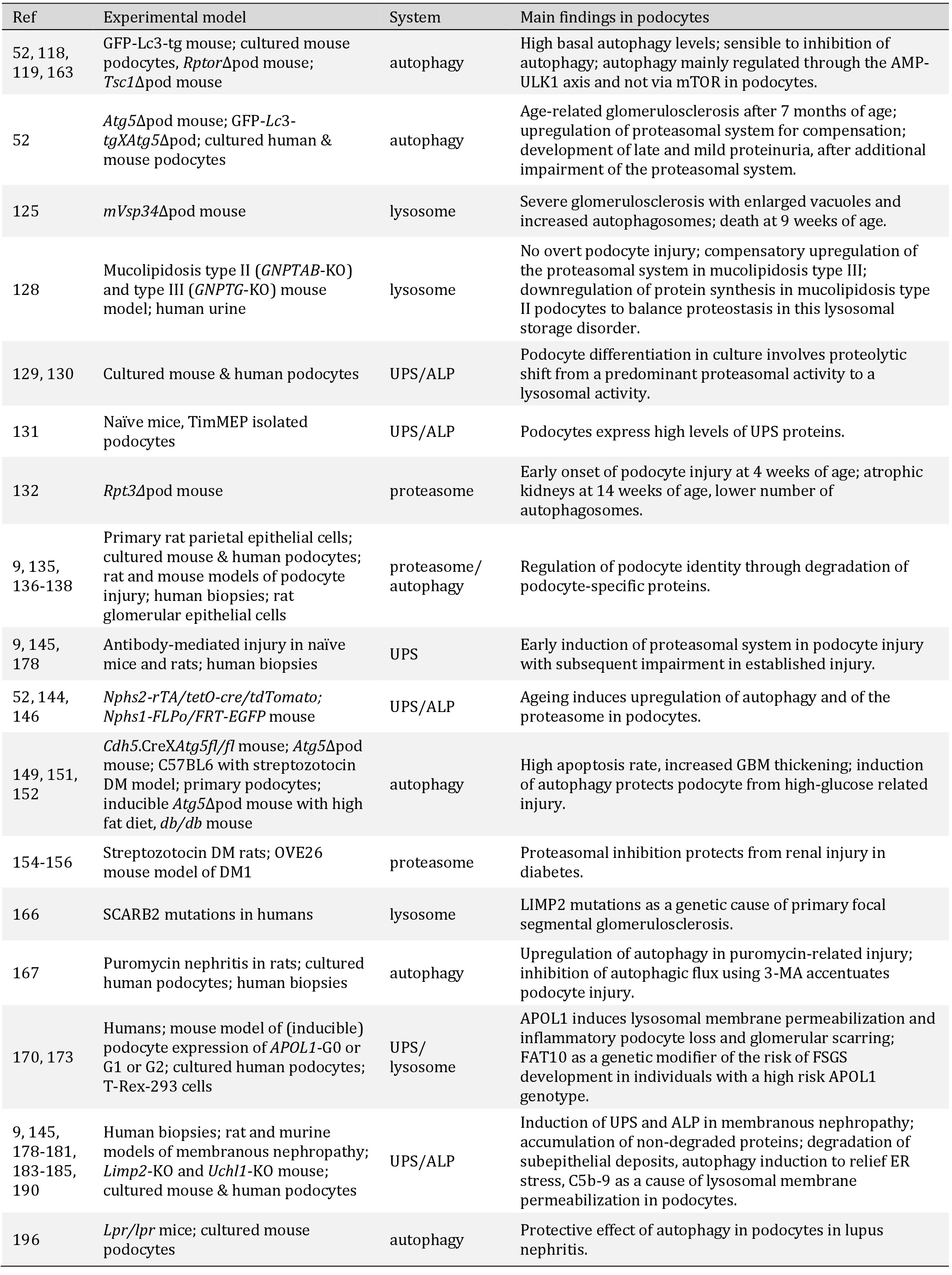
Overview of reviewed findings in selected publications. Abbreviations: Ref = reference number; UPS = ubiquitin proteasomal system, ALP = autophagosomal lysosomal pathway, ∆pod = podocyte specific deletion, ER = endoplasmic reticulum; DM = diabetes mellitus
LH and CMS are supported by the Deutsche Forschungsgesellschaft (DFG grants to CMS ME2180/10-1 and project B3 of the SFB1192).
The authors declare that no conflict of interest exists.
| 1 Meyer-Schwesinger C, Huber T: Glomerular Cell Biology and Podocytopathies, in Yu ASL (ed): Brenner + Rector's The Kidney, 11th ed. Elsevier, Philadelphia, 2020, pp 115-132. | ||||
| 2 Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K: The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med 2008;14:290-298. https://doi.org/10.1038/nm1731 |
||||
| 3 Wang D, Dai C, Li Y, Liu Y: Canonical Wnt/beta-catenin signaling mediates transforming growth factor-beta1-driven podocyte injury and proteinuria. Kidney Int 2011;80:1159-1169. https://doi.org/10.1038/ki.2011.255 |
||||
| 4 Kato H, Gruenwald A, Suh JH, Miner JH, Barisoni-Thomas L, Taketo MM, Faul C, Millar SE, Holzman LB, Susztak K: Wnt/beta-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J Biol Chem 2011;286:26003-26015. https://doi.org/10.1074/jbc.M111.223164 |
||||
| 5 Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y: Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol 2009;20:1997-2008. https://doi.org/10.1681/ASN.2009010019 |
||||
| 6 Shkreli M, Sarin KY, Pech MF, Papeta N, Chang W, Brockman SA, Cheung P, Lee E, Kuhnert F, Olson JL, Kuo CJ, Gharavi AG, D'Agati VD, Artandi SE: Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat Med 2011;18:111-119. https://doi.org/10.1038/nm.2550 |
||||
| 7 Grahammer F, Wanner N, Huber TB: mTOR controls kidney epithelia in health and disease. Nephrol Dial Transplant 2014;29:i9-i18. https://doi.org/10.1093/ndt/gft491 |
||||
| 8 Imasawa T, Rossignol R: Podocyte energy metabolism and glomerular diseases. Int J Biochem Cell Biol 2013;45:2109-2118. https://doi.org/10.1016/j.biocel.2013.06.013 |
||||
| 9 Beeken M, Lindenmeyer MT, Blattner SM, Radon V, Oh J, Meyer TN, Hildebrand D, Schluter H, Reinicke AT, Knop JH, Vivekanandan-Giri A, Munster S, Sachs M, Wiech T, Pennathur S, Cohen CD, Kretzler M, Stahl RA, Meyer-Schwesinger C: Alterations in the ubiquitin proteasome system in persistent but not reversible proteinuric diseases. J Am Soc Nephrol 2014;25:2511-2525. https://doi.org/10.1681/ASN.2013050522 |
||||
| 10 Mitch WE, Goldberg AL: Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med 1996;335:1897-1905. https://doi.org/10.1056/NEJM199612193352507 |
||||
| 11 Lilienbaum A: Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol 2013;4:1-26. | ||||
| 12 Mevissen TET, Komander D: Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev Biochem 2017;86:159-192. https://doi.org/10.1146/annurev-biochem-061516-044916 |
||||
| 13 Komander D, Clague MJ, Urbe S: Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009;10:550-563. https://doi.org/10.1038/nrm2731 |
||||
| 14 Yu H, Singh Gautam AK, Wilmington SR, Wylie D, Martinez-Fonts K, Kago G, Warburton M, Chavali S, Inobe T, Finkelstein IJ, Babu MM, Matouschek A: Conserved Sequence Preferences Contribute to Substrate Recognition by the Proteasome. J Biol Chem 2016;291:14526-14539. https://doi.org/10.1074/jbc.M116.727578 |
||||
| 15 Kisselev AF, Akopian TN, Woo KM, Goldberg AL: The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J Biol Chem 1999;274:3363-3371. https://doi.org/10.1074/jbc.274.6.3363 |
||||
| 16 Murata S, Takahama Y, Kasahara M, Tanaka K: The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat Immunol 2018;19:923-931. https://doi.org/10.1038/s41590-018-0186-z |
||||
| 17 Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M: Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012;148:727-738. https://doi.org/10.1016/j.cell.2011.12.030 |
||||
| 18 Meyer-Schwesinger C: The ubiquitin proteasome system in kidney physiology and disease. Nat Rev Nephrol 2019;15:393-411. https://doi.org/10.1038/s41581-019-0148-1 |
||||
| 19 De Duve C: The lysosome. Sci Am 1963;208:64-72. https://doi.org/10.1038/scientificamerican0563-64 |
||||
| 20 Mijaljica D, Prescott M, Devenish RJ: Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 2011;7:673-682. https://doi.org/10.4161/auto.7.7.14733 |
||||
| 21 Kaushik S, Cuervo AM: The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 2018;19:365-381. https://doi.org/10.1038/s41580-018-0001-6 |
||||
| 22 Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y: Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009;10:458-467. https://doi.org/10.1038/nrm2708 |
||||
| 23 Meyer-Schwesinger C: Lysosome function in glomerular health and disease. Cell Tissue Res 2021; DOI: 10.1007/s00441-020-03375-7. https://doi.org/10.1007/s00441-020-03375-7 |
||||
| 24 Zaffagnini G, Martens S: Mechanisms of Selective Autophagy. J Mol Biol 2016;428:1714-1724. https://doi.org/10.1016/j.jmb.2016.02.004 |
||||
| 25 Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT: Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008;182:685-701. https://doi.org/10.1083/jcb.200803137 |
||||
| 26 Shpilka T, Elazar Z: Lipid droplets regulate autophagosome biogenesis. Autophagy 2015;11:2130-2131. https://doi.org/10.1080/15548627.2015.1093719 |
||||
| 27 Shpilka T, Welter E, Borovsky N, Amar N, Mari M, Reggiori F, Elazar Z: Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J 2015;34:2117-2131. https://doi.org/10.15252/embj.201490315 |
||||
| 28 Hurley JH, Young LN: Mechanisms of Autophagy Initiation. Annu Rev Biochem 2017;86:225-244. https://doi.org/10.1146/annurev-biochem-061516-044820 |
||||
| 29 Suzuki K, Kubota Y, Sekito T, Ohsumi Y: Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007;12:209-218. https://doi.org/10.1111/j.1365-2443.2007.01050.x |
||||
| 30 Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, Teriete P, Asara JM, Turk BE, Cosford ND, Shaw RJ: Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 2015;59:285-297. https://doi.org/10.1016/j.molcel.2015.05.031 |
||||
| 31 Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL: ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013;15:741-750. https://doi.org/10.1038/ncb2757 |
||||
| 32 Mizushima N, Yoshimori T, Levine B: Methods in mammalian autophagy research. Cell 2010;140:313-326. https://doi.org/10.1016/j.cell.2010.01.028 |
||||
| 33 Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ: SNARE proteins are required for macroautophagy. Cell 2011;146:290-302. https://doi.org/10.1016/j.cell.2011.06.022 |
||||
| 34 Ohsumi Y: Historical landmarks of autophagy research. Cell Res 2014;24:9-23. https://doi.org/10.1038/cr.2013.169 |
||||
| 35 Finley D: Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 2009;78:477-513. https://doi.org/10.1146/annurev.biochem.78.081507.101607 |
||||
| 36 International Consortium for Systemic Lupus Erythematosus G, Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, et al.: Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 2008;40:204-210. https://doi.org/10.1038/ng.81 |
||||
| 37 Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, Catanese JJ, Xie G, Stahl EA, Chen R, Alfredsson L, Amos CI, Ardlie KG, Consortium B, Barton A, Bowes J, Burtt NP, Chang M, Coblyn J, Costenbader KH, et al.: Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet 2009;41:1313-1318. https://doi.org/10.1038/ng.479 |
||||
| 38 Brehm A, Kruger E: Dysfunction in protein clearance by the proteasome: impact on autoinflammatory diseases. Semin Immunopathol 2015;37:323-333. https://doi.org/10.1007/s00281-015-0486-4 |
||||
| 39 McDermott A, Jacks J, Kessler M, Emanuel PD, Gao L: Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management. Int J Dermatol 2015;54:121-129. https://doi.org/10.1111/ijd.12695 |
||||
| 40 Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, Almeida de Jesus A, Pelletier M, Tsai WL, Remmers EF, Kardava L, Hill S, Kim H, Lachmann HJ, Megarbane A, Chae JJ, Brady J, et al.: Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 2015;125:4196-4211. https://doi.org/10.1172/JCI81260 |
||||
| 41 Korolchuk VI, Menzies FM, Rubinsztein DC: Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010;584:1393-1398. https://doi.org/10.1016/j.febslet.2009.12.047 |
||||
| 42 Bustamante HA, Gonzalez AE, Cerda-Troncoso C, Shaughnessy R, Otth C, Soza A, Burgos PV: Interplay Between the Autophagy-Lysosomal Pathway and the Ubiquitin-Proteasome System: A Target for Therapeutic Development in Alzheimer's Disease. Front Cell Neurosci 2018;12:126. https://doi.org/10.3389/fncel.2018.00126 |
||||
| 43 B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A: The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013;41:7683-7699. https://doi.org/10.1093/nar/gkt563 |
||||
| 44 Wei Y, Pattingre S, Sinha S, Bassik M, Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008;30:678-688. https://doi.org/10.1016/j.molcel.2008.06.001 |
||||
| 45 Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM: DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006;126:121-134. https://doi.org/10.1016/j.cell.2006.05.034 |
||||
| 46 Zhao B, Qiang L, Joseph J, Kalyanaraman B, Viollet B, He YY: Mitochondrial dysfunction activates the AMPK signaling and autophagy to promote cell survival. Genes Dis 2016;3:82-87. https://doi.org/10.1016/j.gendis.2015.12.002 |
||||
| 47 Gomes LC, Di Benedetto G, Scorrano L: During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 2011;13:589-598. https://doi.org/10.1038/ncb2220 |
||||
| 48 Ugun-Klusek A, Tatham MH, Elkharaz J, Constantin-Teodosiu D, Lawler K, Mohamed H, Paine SM, Anderson G, John Mayer R, Lowe J, Ellen Billett E, Bedford L: Continued 26S proteasome dysfunction in mouse brain cortical neurons impairs autophagy and the Keap1-Nrf2 oxidative defence pathway. Cell Death Dis 2017;8:e2531. https://doi.org/10.1038/cddis.2016.443 |
||||
| 49 Park C, Cuervo AM: Selective autophagy: talking with the UPS. Cell Biochem Biophys 2013;67:3-13. https://doi.org/10.1007/s12013-013-9623-7 |
||||
| 50 Korolchuk VI, Menzies FM, Rubinsztein DC: A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 2009;5:862-863. https://doi.org/10.4161/auto.8840 |
||||
| 51 Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF: p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 2016;21:29. https://doi.org/10.1186/s11658-016-0031-z |
||||
| 52 Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstadt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB: Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 2010;120:1084-1096. https://doi.org/10.1172/JCI39492 |
||||
| 53 Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, Varghese J, Wood N, Wightman M, Osborne G, Bates GP, Glickman MH, Trost M, Knebel A, Marchesi F, Kurz T: UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016;166:935-949. https://doi.org/10.1016/j.cell.2016.07.001 |
||||
| 54 Gao Z, Gammoh N, Wong PM, Erdjument-Bromage H, Tempst P, Jiang X: Processing of autophagic protein LC3 by the 20S proteasome. Autophagy 2010;6:126-137. https://doi.org/10.4161/auto.6.1.10928 |
||||
| 55 Piotrowski J, Beal R, Hoffman L, Wilkinson KD, Cohen RE, Pickart CM: Inhibition of the 26 S proteasome by polyubiquitin chains synthesized to have defined lengths. J Biol Chem 1997;272:23712-23721. https://doi.org/10.1074/jbc.272.38.23712 |
||||
| 56 Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, Yao TP: Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol Cell 2013;51:819-828. https://doi.org/10.1016/j.molcel.2013.08.016 |
||||
| 57 Nanduri P, Hao R, Fitzpatrick T, Yao TP: Chaperone-mediated 26S proteasome remodeling facilitates free K63 ubiquitin chain production and aggresome clearance. J Biol Chem 2015;290:9455-9464. https://doi.org/10.1074/jbc.M114.627950 |
||||
| 58 Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM: Degradation of alpha-synuclein by proteasome. J Biol Chem 1999;274:33855-33858. https://doi.org/10.1074/jbc.274.48.33855 |
||||
| 59 Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC: Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003;278:25009-25013. https://doi.org/10.1074/jbc.M300227200 |
||||
| 60 Bustamante HA, Rivera-Dictter A, Cavieres VA, Munoz VC, Gonzalez A, Lin Y, Mardones GA, Burgos PV: Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degradation in human neuroglioma cells. PLoS One 2013;8:e83096. https://doi.org/10.1371/journal.pone.0083096 |
||||
| 61 Yang C, Cai CZ, Song JX, Tan JQ, Durairajan SSK, Iyaswamy A, Wu MY, Chen LL, Yue Z, Li M, Lu JH: NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017;13:2028-2040. https://doi.org/10.1080/15548627.2017.1379633 |
||||
| 62 Gonzalez AE, Munoz VC, Cavieres VA, Bustamante HA, Cornejo VH, Januario YC, Gonzalez I, Hetz C, daSilva LL, Rojas-Fernandez A, Hay RT, Mardones GA, Burgos PV: Autophagosomes cooperate in the degradation of intracellular C-terminal fragments of the amyloid precursor protein via the MVB/lysosomal pathway. FASEB J 2017;31:2446-2459. https://doi.org/10.1096/fj.201600713R |
||||
| 63 Xiong Q, Li W, Li P, Yang M, Wu C, Eichinger L: The Role of ATG16 in Autophagy and The Ubiquitin Proteasome System. Cells 2018;8:2. https://doi.org/10.3390/cells8010002 |
||||
| 64 Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, Chen RH: Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol Cell 2016;61:84-97. https://doi.org/10.1016/j.molcel.2015.11.001 |
||||
| 65 Khaminets A, Behl C, Dikic I: Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol 2016;26:6-16. https://doi.org/10.1016/j.tcb.2015.08.010 |
||||
| 66 Lu K, Psakhye I, Jentsch S: Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 2014;158:549-563. https://doi.org/10.1016/j.cell.2014.05.048 |
||||
| 67 Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S: Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife 2015;4:e08941. https://doi.org/10.7554/eLife.08941 |
||||
| 68 Hofmann K, Falquet L: A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem Sci 2001;26:347-350. https://doi.org/10.1016/S0968-0004(01)01835-7 |
||||
| 69 Lu K, den Brave F, Jentsch S: Receptor oligomerization guides pathway choice between proteasomal and autophagic degradation. Nat Cell Biol 2017;19:732-739. https://doi.org/10.1038/ncb3531 |
||||
| 70 Lu K, den Brave F, Jentsch S: Pathway choice between proteasomal and autophagic degradation. Autophagy 2017;13:1799-1800. https://doi.org/10.1080/15548627.2017.1358851 |
||||
| 71 Zhou J, Zhang Y, Qi J, Chi Y, Fan B, Yu JQ, Chen Z: E3 ubiquitin ligase CHIP and NBR1-mediated selective autophagy protect additively against proteotoxicity in plant stress responses. PLoS Genet 2014;10:e1004116. https://doi.org/10.1371/journal.pgen.1004116 |
||||
| 72 Kriegenburg F, Jakopec V, Poulsen EG, Nielsen SV, Roguev A, Krogan N, Gordon C, Fleig U, Hartmann-Petersen R: A chaperone-assisted degradation pathway targets kinetochore proteins to ensure genome stability. PLoS Genet 2014;10:e1004140. https://doi.org/10.1371/journal.pgen.1004140 |
||||
| 73 Cohen-Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT, Ciechanover A: p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U S A 2016;113:E7490-E7499. https://doi.org/10.1073/pnas.1615455113 |
||||
| 74 Grice GL, Nathan JA: The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci 2016;73:3497-3506. https://doi.org/10.1007/s00018-016-2255-5 |
||||
| 75 Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T: A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009;33:505-516. https://doi.org/10.1016/j.molcel.2009.01.020 |
||||
| 76 Waters S, Marchbank K, Solomon E, Whitehouse C, Gautel M: Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett 2009;583:1846-1852. https://doi.org/10.1016/j.febslet.2009.04.049 |
||||
| 77 Kirkin V, McEwan DG, Novak I, Dikic I: A role for ubiquitin in selective autophagy. Mol Cell 2009;34:259-269. https://doi.org/10.1016/j.molcel.2009.04.026 |
||||
| 78 Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I: Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008;453:481-488. https://doi.org/10.1038/nature06926 |
||||
| 79 Yu H, Matouschek A: Recognition of Client Proteins by the Proteasome. Annu Rev Biophys 2017;46:149-173. https://doi.org/10.1146/annurev-biophys-070816-033719 |
||||
| 80 Verma R, Aravind L, Oania R, McDonald WH, Yates JR, 3rd, Koonin EV, Deshaies RJ: Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002;298:611-615. https://doi.org/10.1126/science.1075898 |
||||
| 81 Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW: Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004;24:8055-8068. https://doi.org/10.1128/MCB.24.18.8055-8068.2004 |
||||
| 82 N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ: PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009;10:173-179. https://doi.org/10.1038/embor.2008.238 |
||||
| 83 Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T: p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005;171:603-614. https://doi.org/10.1083/jcb.200507002 |
||||
| 84 Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, et al.: Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007;131:1149-1163. https://doi.org/10.1016/j.cell.2007.10.035 |
||||
| 85 Peng H, Yang J, Li G, You Q, Han W, Li T, Gao D, Xie X, Lee BH, Du J, Hou J, Zhang T, Rao H, Huang Y, Li Q, Zeng R, Hui L, Wang H, Xia Q, Zhang X, et al.: Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res 2017;27:657-674. https://doi.org/10.1038/cr.2017.40 |
||||
| 86 Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T: p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010;285:22576-22591. https://doi.org/10.1074/jbc.M110.118976 |
||||
| 87 Vadlamudi RK, Joung I, Strominger JL, Shin J: p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 1996;271:20235-20237. https://doi.org/10.1074/jbc.271.34.20235 |
||||
| 88 Itakura E, Mizushima N: p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 2011;192:17-27. https://doi.org/10.1083/jcb.201009067 |
||||
| 89 Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T: p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131-24145. https://doi.org/10.1074/jbc.M702824200 |
||||
| 90 Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC: Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 2011;20:1726-1737. https://doi.org/10.1093/hmg/ddr048 |
||||
| 91 Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W: PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010;12:119-131. https://doi.org/10.1038/ncb2012 |
||||
| 92 Eiyama A, Okamoto K: PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol 2015;33:95-101. https://doi.org/10.1016/j.ceb.2015.01.002 |
||||
| 93 Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ: The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015;524:309-314. https://doi.org/10.1038/nature14893 |
||||
| 94 Fusco C, Micale L, Egorov M, Monti M, D'Addetta EV, Augello B, Cozzolino F, Calcagni A, Fontana A, Polishchuk RS, Didelot G, Reymond A, Pucci P, Merla G: The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS One 2012;7:e40440. https://doi.org/10.1371/journal.pone.0040440 |
||||
| 95 Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, Wada K: Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet 2003;12:1945-1958. https://doi.org/10.1093/hmg/ddg211 |
||||
| 96 Burgos PV, Mardones GA, Rojas AL, daSilva LL, Prabhu Y, Hurley JH, Bonifacino JS: Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell 2010;18:425-436. https://doi.org/10.1016/j.devcel.2010.01.015 |
||||
| 97 Prabhu Y, Burgos PV, Schindler C, Farias GG, Magadan JG, Bonifacino JS: Adaptor protein 2-mediated endocytosis of the beta-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol Biol Cell 2012;23:2339-2351. https://doi.org/10.1091/mbc.e11-11-0944 |
||||
| 98 Collins GA, Goldberg AL: The Logic of the 26S Proteasome. Cell 2017;169:792-806. https://doi.org/10.1016/j.cell.2017.04.023 |
||||
| 99 Srinivasan V, Bruelle C, Scifo E, Pham DD, Soliymani R, Lalowski M, Lindholm D: Dynamic Interaction of USP14 with the Chaperone HSC70 Mediates Crosstalk between the Proteasome, ER Signaling, and Autophagy. iScience 2020;23:100790. https://doi.org/10.1016/j.isci.2019.100790 |
||||
| 100 Xu D, Shan B, Sun H, Xiao J, Zhu K, Xie X, Li X, Liang W, Lu X, Qian L, Yuan J: USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev 2016;30:1718-1730. https://doi.org/10.1101/gad.285122.116 |
||||
| 101 Castano JG, Mahillo E, Arizti P, Arribas J: Phosphorylation of C8 and C9 subunits of the multicatalytic proteinase by casein kinase II and identification of the C8 phosphorylation sites by direct mutagenesis. Biochemistry 1996;35:3782-3789. https://doi.org/10.1021/bi952540s |
||||
| 102 Mason GG, Hendil KB, Rivett AJ: Phosphorylation of proteasomes in mammalian cells. Identification of two phosphorylated subunits and the effect of phosphorylation on activity. Eur J Biochem 1996;238:453-462. https://doi.org/10.1111/j.1432-1033.1996.0453z.x |
||||
| 103 Kanki T, Kurihara Y, Jin X, Goda T, Ono Y, Aihara M, Hirota Y, Saigusa T, Aoki Y, Uchiumi T, Kang D: Casein kinase 2 is essential for mitophagy. EMBO Rep 2013;14:788-794. https://doi.org/10.1038/embor.2013.114 |
||||
| 104 Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN: Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem 2009;284:26655-26665. https://doi.org/10.1074/jbc.M109.021956 |
||||
| 105 Jarome TJ, Ferrara NC, Kwapis JL, Helmstetter FJ: CaMKII regulates proteasome phosphorylation and activity and promotes memory destabilization following retrieval. Neurobiol Learn Mem 2016;128:103-109. https://doi.org/10.1016/j.nlm.2016.01.001 |
||||
| 106 Li X, Wu XQ, Deng R, Li DD, Tang J, Chen WD, Chen JH, Ji J, Jiao L, Jiang S, Yang F, Feng GK, Senthilkumar R, Yue F, Zhang HL, Wu RY, Yu Y, Xu XL, Mai J, Li ZL, et al.: CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat Commun 2017;8:1159. https://doi.org/10.1038/s41467-017-01272-2 |
||||
| 107 Satoh K, Sasajima H, Nyoumura KI, Yokosawa H, Sawada H: Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 2001;40:314-319. https://doi.org/10.1021/bi001815n |
||||
| 108 Lokireddy S, Kukushkin NV, Goldberg AL: cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A 2015;112:E7176-7185. https://doi.org/10.1073/pnas.1522332112 |
||||
| 109 Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK: The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci U S A 2009;106:17049-17054. https://doi.org/10.1073/pnas.0903316106 |
||||
| 110 Torres-Quiroz F, Filteau M, Landry CR: Feedback regulation between autophagy and PKA. Autophagy 2015;11:1181-1183. https://doi.org/10.1080/15548627.2015.1055440 |
||||
| 111 Asano S, Fukuda Y, Beck F, Aufderheide A, Forster F, Danev R, Baumeister W: Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 2015;347:439-442. https://doi.org/10.1126/science.1261197 |
||||
| 112 Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, Ciechanover A: The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res 2016;26:869-885. https://doi.org/10.1038/cr.2016.86 |
||||
| 113 Onodera J, Ohsumi Y: Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem 2005;280:31582-31586. https://doi.org/10.1074/jbc.M506736200 |
||||
| 114 Lenoir O, Tharaux PL, Huber TB: Autophagy in kidney disease and aging: lessons from rodent models. Kidney Int 2016;90:950-964. https://doi.org/10.1016/j.kint.2016.04.014 |
||||
| 115 Cybulsky AV: The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int 2013;84:25-33. https://doi.org/10.1038/ki.2012.390 |
||||
| 116 Cybulsky AV: Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature reviews Nephrology 2017;13:681-696. https://doi.org/10.1038/nrneph.2017.129 |
||||
| 117 Wanner N, Hartleben B, Herbach N, Goedel M, Stickel N, Zeiser R, Walz G, Moeller MJ, Grahammer F, Huber TB: Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol 2014;25:707-716. https://doi.org/10.1681/ASN.2013050452 |
||||
| 118 Fang L, Li X, Luo Y, He W, Dai C, Yang J: Autophagy inhibition induces podocyte apoptosis by activating the pro-apoptotic pathway of endoplasmic reticulum stress. Exp Cell Res 2014;322:290-301. https://doi.org/10.1016/j.yexcr.2014.01.001 |
||||
| 119 Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y: In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004;15:1101-1111. https://doi.org/10.1091/mbc.e03-09-0704 |
||||
| 120 Miller F, Palade GE: Lytic Activities in Renal Protein Absorption Droplets. An Electron Microscopical Cytochemical Study. J Cell Biol 1964;23:519-552. https://doi.org/10.1083/jcb.23.3.519 |
||||
| 121 Lovett DH, Ryan JL, Kashgarian M, Sterzel RB: Lysosomal enzymes in glomerular cells of the rat. Am J Pathol 1982;107:161-166. | ||||
| 122 Yokota S, Tsuji H, Kato K: Immunocytochemical localization of cathepsin D in lysosomes of cortical collecting tubule cells of the rat kidney. J Histochem Cytochem 1985;33:191-200. https://doi.org/10.1177/33.3.2579120 |
||||
| 123 Herman PK, Emr SD: Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol Cell Biol 1990;10:6742-6754. https://doi.org/10.1128/mcb.10.12.6742-6754.1990 |
||||
| 124 Itakura E, Kishi C, Inoue K, Mizushima N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008;19:5360-5372. https://doi.org/10.1091/mbc.e08-01-0080 |
||||
| 125 Chen J, Chen MX, Fogo AB, Harris RC, Chen JK: mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. J Am Soc Nephrol 2013;24:198-207. https://doi.org/10.1681/ASN.2012010101 |
||||
| 126 Velho RV, Harms FL, Danyukova T, Ludwig NF, Friez MJ, Cathey SS, Filocamo M, Tappino B, Gunes N, Tuysuz B, Tylee KL, Brammeier KL, Heptinstall L, Oussoren E, van der Ploeg AT, Petersen C, Alves S, Saavedra GD, Schwartz IV, Muschol N, et al.: The lysosomal storage disorders mucolipidosis type II, type III alpha/beta and type III gamma: Update on GNPTAB and GNPTG mutations. Hum Mutat 2019;40:842-864. https://doi.org/10.1002/humu.23748 |
||||
| 127 Kollmann K, Pohl S, Marschner K, Encarnacao M, Sakwa I, Tiede S, Poorthuis BJ, Lubke T, Muller-Loennies S, Storch S, Braulke T: Mannose phosphorylation in health and disease. Eur J Cell Biol 2010;89:117-123. https://doi.org/10.1016/j.ejcb.2009.10.008 |
||||
| 128 Sachs W, Sachs M, Kruger E, Zielinski S, Kretz O, Huber TB, Baranowsky A, Westermann LM, Voltolini Velho R, Ludwig NF, Yorgan TA, Di Lorenzo G, Kollmann K, Braulke T, Schwartz IV, Schinke T, Danyukova T, Pohl S, Meyer-Schwesinger C: Distinct Modes of Balancing Glomerular Cell Proteostasis in Mucolipidosis Type II and III Prevent Proteinuria. J Am Soc Nephrol 2020;31:1796-1814. https://doi.org/10.1681/ASN.2019090960 |
||||
| 129 Rinschen MM, Schroeter CB, Koehler S, Ising C, Schermer B, Kann M, Benzing T, Brinkkoetter PT: Quantitative deep mapping of the cultured podocyte proteome uncovers shifts in proteostatic mechanisms during differentiation. Am J Physiol Cell Physiol 2016;311:C404-417. https://doi.org/10.1152/ajpcell.00121.2016 |
||||
| 130 Schroeter CB, Koehler S, Kann M, Schermer B, Benzing T, Brinkkoetter PT, Rinschen MM: Protein half-life determines expression of proteostatic networks in podocyte differentiation. FASEB J 2018;32:4696-4713. https://doi.org/10.1096/fj.201701307R |
||||
| 131 Hatje F, Wedekind U, Sachs W, Loreth D, Reichelt J, Demir F, Kosub C, Heintz L, Tomas N, Huber T, Skuza S, Sachs M, Zielinski S, Rinschen M, Meyer-Schwesinger C: Tripartite Separation of Glomerular Cell-Types and Proteomes From Reporter-Free Mice. J Am Soc Nephrol 2021;32:2175-2193. https://doi.org/10.1681/ASN.2020091346 |
||||
| 132 Makino SI, Shirata N, Oliva Trejo JA, Yamamoto-Nonaka K, Yamada H, Miyake T, Mori K, Nakagawa T, Tashiro Y, Yamashita H, Yanagita M, Takahashi R, Asanuma K: Impairment of Proteasome Function in Podocytes Leads to CKD. J Am Soc Nephrol 2021;32:597-613. https://doi.org/10.1681/ASN.2019101025 |
||||
| 133 Kann M, Ettou S, Jung YL, Lenz MO, Taglienti ME, Park PJ, Schermer B, Benzing T, Kreidberg JA: Genome-Wide Analysis of Wilms' Tumor 1-Controlled Gene Expression in Podocytes Reveals Key Regulatory Mechanisms. J Am Soc Nephrol 2015;26:2097-2104. https://doi.org/10.1681/ASN.2014090940 |
||||
| 134 Zhou L, Li Y, He W, Zhou D, Tan RJ, Nie J, Hou FF, Liu Y: Mutual antagonism of Wilms' tumor 1 and beta-catenin dictates podocyte health and disease. J Am Soc Nephrol 2015;26:677-691. https://doi.org/10.1681/ASN.2013101067 |
||||
| 135 Rinschen MM, Bharill P, Wu X, Kohli P, Reinert MJ, Kretz O, Saez I, Schermer B, Hohne M, Bartram MP, Aravamudhan S, Brooks BR, Vilchez D, Huber TB, Muller RU, Kruger M, Benzing T: The ubiquitin ligase Ubr4 controls stability of podocin/MEC-2 supercomplexes. Hum Mol Genet 2016;25:1328-1344. https://doi.org/10.1093/hmg/ddw016 |
||||
| 136 Guhr SS, Sachs M, Wegner A, Becker JU, Meyer TN, Kietzmann L, Schlossarek S, Carrier L, Braig M, Jat PS, Stahl RA, Meyer-Schwesinger C: The expression of podocyte-specific proteins in parietal epithelial cells is regulated by protein degradation. Kidney Int 2013;84:532-544. https://doi.org/10.1038/ki.2013.115 |
||||
| 137 Cybulsky AV, Takano T, Papillon J, Bijian K, Guillemette J, Kennedy CR: Glomerular epithelial cell injury associated with mutant alpha-actinin-4. Am J Physiol Renal Physiol 2009;297:F987-995. https://doi.org/10.1152/ajprenal.00055.2009 |
||||
| 138 Serrano-Perez MC, Tilley FC, Nevo F, Arrondel C, Sbissa S, Martin G, Tory K, Antignac C, Mollet G: Endoplasmic reticulum-retained podocin mutants are massively degraded by the proteasome. J Biol Chem 2018;293:4122-4133. https://doi.org/10.1074/jbc.RA117.001159 |
||||
| 139 Yee A, Papillon J, Guillemette J, Kaufman DR, Kennedy CRJ, Cybulsky AV: Proteostasis as a therapeutic target in glomerular injury associated with mutant alpha-actinin-4. Am J Physiol Renal Physiol 2018;315:F954-F966. https://doi.org/10.1152/ajprenal.00082.2018 |
||||
| 140 Tossidou I, Teng B, Drobot L, Meyer-Schwesinger C, Worthmann K, Haller H, Schiffer M: CIN85/RukL is a novel binding partner of nephrin and podocin and mediates slit diaphragm turnover in podocytes. J Biol Chem 2010;285:25285-25295. https://doi.org/10.1074/jbc.M109.087239 |
||||
| 141 Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, Grunwald T, Sellin L: beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci U S A 2006;103:14110-14115. https://doi.org/10.1073/pnas.0602587103 |
||||
| 142 Tossidou I, Himmelseher E, Teng B, Haller H, Schiffer M: SUMOylation determines turnover and localization of nephrin at the plasma membrane. Kidney Int 2014;86:1161-1173. https://doi.org/10.1038/ki.2014.198 |
||||
| 143 Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P: Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 2006;8:485-491. https://doi.org/10.1038/ncb1400 |
||||
| 144 Buvall L, Rashmi P, Lopez-Rivera E, Andreeva S, Weins A, Wallentin H, Greka A, Mundel P: Proteasomal degradation of Nck1 but not Nck2 regulates RhoA activation and actin dynamics. Nat Commun 2013;4:2863. https://doi.org/10.1038/ncomms3863 |
||||
| 145 Radon V, Czesla M, Reichelt J, Fehlert J, Hammel A, Rosendahl A, Knop JH, Wiech T, Wenzel UO, Sachs M, Reinicke AT, Stahl RAK, Meyer-Schwesinger C: Ubiquitin C-Terminal Hydrolase L1 is required for regulated protein degradation through the ubiquitin proteasome system in kidney. Kidney Int 2018;93:110-127. https://doi.org/10.1016/j.kint.2017.05.016 |
||||
| 146 Wang Y, Eng DG, Kaverina NV, Loretz CJ, Koirala A, Akilesh S, Pippin JW, Shankland SJ: Global transcriptomic changes occur in aged mouse podocytes. Kidney Int 2020;98:1160-1173. https://doi.org/10.1016/j.kint.2020.05.052 |
||||
| 147 Kitada M, Ogura Y, Monno I, Koya D: Regulating Autophagy as a Therapeutic Target for Diabetic Nephropathy. Curr Diab Rep 2017;17:53. https://doi.org/10.1007/s11892-017-0879-y |
||||
| 148 Yasuda-Yamahara M, Kume S, Tagawa A, Maegawa H, Uzu T: Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy 2015;11:2385-2386. https://doi.org/10.1080/15548627.2015.1115173 |
||||
| 149 Lenoir O, Jasiek M, Henique C, Guyonnet L, Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A, Masse JM, Souyri M, Huber TB, Tharaux PL: Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015;11:1130-1145. https://doi.org/10.1080/15548627.2015.1049799 |
||||
| 150 Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K, Kim EH, Haneda M, Kajiwara N, Hayashi K, Ohashi H, Ugi S, Maegawa H, Uzu T: Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016;65:755-767. https://doi.org/10.2337/db15-0473 |
||||
| 151 Huang SS, Ding DF, Chen S, Dong CL, Ye XL, Yuan YG, Feng YM, You N, Xu JR, Miao H, You Q, Lu X, Lu YB: Resveratrol protects podocytes against apoptosis via stimulation of autophagy in a mouse model of diabetic nephropathy. Sci Rep 2017;7:45692. https://doi.org/10.1038/srep45692 |
||||
| 152 Li X, Zhu Q, Zheng R, Yan J, Wei M, Fan Y, Deng Y, Zhong Y: Puerarin Attenuates Diabetic Nephropathy by Promoting Autophagy in Podocytes. Front Physiol 2020;11:73. https://doi.org/10.3389/fphys.2020.00073 |
||||
| 153 Yoshibayashi M, Kume S, Yasuda-Yamahara M, Yamahara K, Takeda N, Osawa N, Chin-Kanasaki M, Nakae Y, Yokoi H, Mukoyama M, Asanuma K, Maegawa H, Araki SI: Protective role of podocyte autophagy against glomerular endothelial dysfunction in diabetes. Biochem Biophys Res Commun 2020;525:319-325. https://doi.org/10.1016/j.bbrc.2020.02.088 |
||||
| 154 Luo ZF, Qi W, Feng B, Mu J, Zeng W, Guo YH, Pang Q, Ye ZL, Liu L, Yuan FH: Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sci 2011;88:512-520. https://doi.org/10.1016/j.lfs.2010.12.023 |
||||
| 155 Cui W, Li B, Bai Y, Miao X, Chen Q, Sun W, Tan Y, Luo P, Zhang C, Zheng S, Epstein PN, Miao L, Cai L: Potential role for Nrf2 activation in the therapeutic effect of MG132 on diabetic nephropathy in OVE26 diabetic mice. Am J Physiol Endocrinol Metab 2013;304:E87-99. https://doi.org/10.1152/ajpendo.00430.2012 |
||||
| 156 Zeng W, Qi W, Mu J, Wei Y, Yang LL, Zhang Q, Wu Q, Tang JY, Feng B: MG132 protects against renal dysfunction by regulating Akt-mediated inflammation in diabetic nephropathy. Sci Rep 2019;9:2049. https://doi.org/10.1038/s41598-018-38425-2 |
||||
| 157 Dimke H, Maezawa Y, Quaggin SE: Crosstalk in glomerular injury and repair. Curr Opin Nephrol Hypertens 2015;24:231-238. https://doi.org/10.1097/MNH.0000000000000117 |
||||
| 158 Njomen E, Tepe JJ: Regulation of Autophagic Flux by the 20S Proteasome. Cell Chem Biol 2019;26:1283-1294.e5. https://doi.org/10.1016/j.chembiol.2019.07.002 |
||||
| 159 Dong C, Zheng H, Huang S, You N, Xu J, Ye X, Zhu Q, Feng Y, You Q, Miao H, Ding D, Lu Y: Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp Cell Res 2015;337:146-159. https://doi.org/10.1016/j.yexcr.2015.04.005 |
||||
| 160 Komander D, Rape M: The ubiquitin code. Annu Rev Biochem 2012;81:203-229. https://doi.org/10.1146/annurev-biochem-060310-170328 |
||||
| 161 Tenno T, Fujiwara K, Tochio H, Iwai K, Morita EH, Hayashi H, Murata S, Hiroaki H, Sato M, Tanaka K, Shirakawa M: Structural basis for distinct roles of Lys63- and Lys48-linked polyubiquitin chains. Genes Cells 2004;9:865-875. https://doi.org/10.1111/j.1365-2443.2004.00780.x |
||||
| 162 Jiang S, Park DW, Gao Y, Ravi S, Darley-Usmar V, Abraham E, Zmijewski JW: Participation of proteasome-ubiquitin protein degradation in autophagy and the activation of AMP-activated protein kinase. Cell Signal 2015;27:1186-1197. https://doi.org/10.1016/j.cellsig.2015.02.024 |
||||
| 163 Bork T, Liang W, Yamahara K, Lee P, Tian Z, Liu S, Schell C, Thedieck K, Hartleben B, Patel K, Tharaux PL, Lenoir O, Huber TB: Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy 2020;16:1932-1948. https://doi.org/10.1080/15548627.2019.1705007 |
||||
| 164 Fogo AB: Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 2015;11:76-87. https://doi.org/10.1038/nrneph.2014.216 |
||||
| 165 Bierzynska A, Soderquest K, Koziell A: Genes and podocytes - new insights into mechanisms of podocytopathy. Front Endocrinol (Lausanne) 2014;5:226. https://doi.org/10.3389/fendo.2014.00226 |
||||
| 166 Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, Lullmann-Rauch R, Blanz J, Zhang KW, Stankovich J, Kalnins RM, Dowling JP, Andermann E, Andermann F, Faldini E, D'Hooge R, Vadlamudi L, Macdonell RA, Hodgson BL, Bayly MA, et al.: Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 2008;82:673-684. https://doi.org/10.1016/j.ajhg.2007.12.019 |
||||
| 167 Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong Y, Zhang C, Zen K, Liu Z: Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol 2014;234:203-213. https://doi.org/10.1002/path.4382 |
||||
| 168 Yang JW, Dettmar AK, Kronbichler A, Gee HY, Saleem M, Kim SH, Shin JI: Recent advances of animal model of focal segmental glomerulosclerosis. Clin Exp Nephrol 2018;22:752-763. https://doi.org/10.1007/s10157-018-1552-8 |
||||
| 169 Genovese G, Friedman DJ, Pollak MR: APOL1 variants and kidney disease in people of recent African ancestry. Nat Rev Nephrol 2013;9:240-244. https://doi.org/10.1038/nrneph.2013.34 |
||||
| 170 Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu CA, Rohacs T, Inoue K, Ishibe S, Saleem MA, Palmer MB, Cuervo AM, Kopp JB, Susztak K: Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017;23:429-438. https://doi.org/10.1038/nm.4287 |
||||
| 171 Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, Jacquet A, Brasseur R, Pays E: Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005;309:469-472. https://doi.org/10.1126/science.1114566 |
||||
| 172 Hipp MS, Kalveram B, Raasi S, Groettrup M, Schmidtke G: FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol Cell Biol 2005;25:3483-3491. https://doi.org/10.1128/MCB.25.9.3483-3491.2005 |
||||
| 173 Zhang JY, Wang M, Tian L, Genovese G, Yan P, Wilson JG, Thadhani R, Mottl AK, Appel GB, Bick AG, Sampson MG, Alper SL, Friedman DJ, Pollak MR: UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci U S A 2018;115:3446-3451. https://doi.org/10.1073/pnas.1716113115 |
||||
| 174 Beck LH, Jr., Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ: M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009;361:11-21. https://doi.org/10.1056/NEJMoa0810457 |
||||
| 175 Tomas NM, Beck LH, Jr., Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, Dolla G, Hoxha E, Helmchen U, Dabert-Gay AS, Debayle D, Merchant M, Klein J, Salant DJ, Stahl RAK, Lambeau G: Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 2014;371:2277-2287. https://doi.org/10.1056/NEJMoa1409354 |
||||
| 176 Beck LH, Jr.: PLA2R and THSD7A: Disparate Paths to the Same Disease? J Am Soc Nephrol 2017;28:2579-2589. https://doi.org/10.1681/ASN.2017020178 |
||||
| 177 Herwig J, Skuza S, Sachs W, Sachs M, Failla AV, Rune G, Meyer TN, Fester L, Meyer-Schwesinger C: Thrombospondin Type 1 Domain-Containing 7A Localizes to the Slit Diaphragm and Stabilizes Membrane Dynamics of Fully Differentiated Podocytes. J Am Soc Nephrol 2019;30:824-839. https://doi.org/10.1681/ASN.2018090941 |
||||
| 178 Meyer-Schwesinger C, Meyer TN, Munster S, Klug P, Saleem M, Helmchen U, Stahl RA: A new role for the neuronal ubiquitin C-terminal hydrolase-L1 (UCH-L1) in podocyte process formation and podocyte injury in human glomerulopathies. J Pathol 2009;217:452-464. https://doi.org/10.1002/path.2446 |
||||
| 179 Rood IM, Merchant ML, Wilkey DW, Zhang T, Zabrouskov V, van der Vlag J, Dijkman HB, Willemsen BK, Wetzels JF, Klein JB, Deegens JK: Increased expression of lysosome membrane protein 2 in glomeruli of patients with idiopathic membranous nephropathy. Proteomics 2015;15:3722-3730. https://doi.org/10.1002/pmic.201500127 |
||||
| 180 Wu CC, Chen JS, Huang CF, Chen CC, Lu KC, Chu P, Sytwu HK, Lin YF: Approaching biomarkers of membranous nephropathy from a murine model to human disease. J Biomed Biotechnol 2011;2011:581928. https://doi.org/10.1155/2011/581928 |
||||
| 181 Singh AK: Proteolytic machinery of glomerular epithelial cells against IgG. Biochem Biophys Res Commun 1992;186:639-644. https://doi.org/10.1016/0006-291X(92)90794-L |
||||
| 182 Borza DB, Zhang JJ, Beck LH, Jr., Meyer-Schwesinger C, Luo W: Mouse models of membranous nephropathy: the road less travelled by. Am J Clin Exp Immunol 2013;2:135-145. | ||||
| 183 Wang L, Hong Q, Lv Y, Feng Z, Zhang X, Wu L, Cui S, Hou K, Su H, Huang Z, Wu D, Chen X: Autophagy can repair endoplasmic reticulum stress damage of the passive Heymann nephritis model as revealed by proteomics analysis. J Proteomics 2012;75:3866-3876. https://doi.org/10.1016/j.jprot.2012.04.016 |
||||
| 184 Liu WJ, Li ZH, Chen XC, Zhao XL, Zhong Z, Yang C, Wu HL, An N, Li WY, Liu HF: Blockage of the lysosome-dependent autophagic pathway contributes to complement membrane attack complex-induced podocyte injury in idiopathic membranous nephropathy. Sci Rep 2017;7:8643. https://doi.org/10.1038/s41598-017-07889-z |
||||
| 185 Kitzler TM, Papillon J, Guillemette J, Wing SS, Cybulsky AV: Complement modulates the function of the ubiquitin-proteasome system and endoplasmic reticulum-associated degradation in glomerular epithelial cells. Biochim Biophys Acta 2012;1823:1007-1016. https://doi.org/10.1016/j.bbamcr.2012.03.001 |
||||
| 186 Meyer-Schwesinger C, Meyer TN, Sievert H, Hoxha E, Sachs M, Klupp EM, Munster S, Balabanov S, Carrier L, Helmchen U, Thaiss F, Stahl RA: Ubiquitin C-terminal hydrolase-l1 activity induces polyubiquitin accumulation in podocytes and increases proteinuria in rat membranous nephropathy. Am J Pathol 2011;178:2044-2057. https://doi.org/10.1016/j.ajpath.2011.01.017 |
||||
| 187 Liu Y, Wu H, Wu J, Wang S, Liu Y, Zhao Z, Zhang X, Li R, Guo M, Zhang Z: Detection of UCH-L1 expression by pre-embedding immunoelectron microscopy with colloidal gold labeling in diseased glomeruli. Ultrastruct Pathol 2008;32:5-9. https://doi.org/10.1080/01913120701829285 |
||||
| 188 Liu Y, Wu J, Wu H, Wang T, Gan H, Zhang X, Liu Y, Li R, Zhao Z, Chen Q, Guo M, Zhang Z: UCH-L1 expression of podocytes in diseased glomeruli and in vitro. J Pathol 2009;217:642-653. https://doi.org/10.1002/path.2511 |
||||
| 189 Sosna J, Voigt S, Mathieu S, Kabelitz D, Trad A, Janssen O, Meyer-Schwesinger C, Schutze S, Adam D: The proteases HtrA2/Omi and UCH-L1 regulate TNF-induced necroptosis. Cell Commun Signal 2013;11:76. https://doi.org/10.1186/1478-811X-11-76 |
||||
| 190 Boisvert NC, Holterman CE, Thibodeau JF, Nasrallah R, Kamto E, Comin CH, da FCL, Carter A, Hebert RL, Gutsol A, Cron GO, Lacoste B, Gray DA, Kennedy CR: Hyperfiltration in Ubiquitin C-Terminal Hydrolase L1 -Deleted Mice. Clin Sci (Lond) 2018 https://doi.org/10.1042/CS20180085 |
||||
| 191 Lohmann F, Sachs M, Meyer TN, Sievert H, Lindenmeyer MT, Wiech T, Cohen CD, Balabanov S, Stahl RA, Meyer-Schwesinger C: UCH-L1 induces podocyte hypertrophy in membranous nephropathy by protein accumulation. Biochim Biophys Acta 2014;1842:945-958. https://doi.org/10.1016/j.bbadis.2014.02.011 |
||||
| 192 Kabuta T, Setsuie R, Mitsui T, Kinugawa A, Sakurai M, Aoki S, Uchida K, Wada K: Aberrant molecular properties shared by familial Parkinson's disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum Mol Genet 2008;17:1482-1496. https://doi.org/10.1093/hmg/ddn037 |
||||
| 193 Schrezenmeier E, Jayne D, Dorner T: Targeting B Cells and Plasma Cells in Glomerular Diseases: Translational Perspectives. J Am Soc Nephrol 2018;29:741-758. https://doi.org/10.1681/ASN.2017040367 |
||||
| 194 Li J, Basler M, Alvarez G, Brunner T, Kirk CJ, Groettrup M: Immunoproteasome inhibition prevents chronic antibody-mediated allograft rejection in renal transplantation. Kidney Int 2018;93:670-680. https://doi.org/10.1016/j.kint.2017.09.023 |
||||
| 195 Li J, Koerner J, Basler M, Brunner T, Kirk CJ, Groettrup M: Immunoproteasome inhibition induces plasma cell apoptosis and preserves kidney allografts by activating the unfolded protein response and suppressing plasma cell survival factors. Kidney Int 2019;95:611-623. https://doi.org/10.1016/j.kint.2018.10.022 |
||||
| 196 Qi YY, Zhou XJ, Cheng FJ, Hou P, Ren YL, Wang SX, Zhao MH, Yang L, Martinez J, Zhang H: Increased autophagy is cytoprotective against podocyte injury induced by antibody and interferon-alpha in lupus nephritis. Ann Rheum Dis 2018;77:1799-1809. https://doi.org/10.1136/annrheumdis-2018-213028 |
||||
| 197 Castellano G, Cafiero C, Divella C, Sallustio F, Gigante M, Pontrelli P, De Palma G, Rossini M, Grandaliano G, Gesualdo L: Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and LMP7 induction in renal tubular epithelial cells. Arthritis Res Ther 2015;17:72. https://doi.org/10.1186/s13075-015-0588-3 |
||||