×
![]()
Corresponding Author: Carlos E. Irarrázabal
Laboratorio de Fisiología Integrativa y Molecular, Universidad de los Andes, S. Carlos Apoquindo 2200-Las Condes, Santiago (Chile)
Tel. +56-2-41291607, Fax +56-2-2141756 , E-Mail cirarrazabal@uandes.cl
Glutathione S-Transferase and Clusterin, New Players in the Ischemic Preconditioning Renal Protection in a Murine Model of Ischemia and Reperfusion
Consuelo Pastena,b Yeimi Herrera-Lunaa Mauricio Lozanoa Jocelyn Roccoa Cristobal Alvaradoc,d Jéssica Liberonae Luis Micheae Carlos E. Irarrázabala,b
aLaboratorio de Fisiología Integrativa y Molecular, Programa de Fisiología, Centro de Investigación e Innovación Biomédica, Universidad de los Andes, Santiago, Chile, bFacultad de Medicina, Universidad de los Andes, Santiago, Chile, cClinical Research Unit, Hospital Las Higueras, Talcahuano, Chile, dDepartment of Basic Sciences, School of Medicine, Universidad Católica de la Santísima Concepción, Concepción, Chile, eInstituto de Ciencias Biomédicas, School of Medicine, Universidad de Chile, Santiago, Chile
Introduction
Acute Kidney Injury (AKI) is defined as a sudden and sustained fall of renal function, which is associated with poor outcomes such as prolonged length of both intensive care and hospital stays, advanced chronic kidney disease (CKD), and death [1]. The kidney is one of the most susceptible organs to ischemia, and renal ischemia/reperfusion represents a leading cause of AKI [2-5]. Ischemia and reperfusion injury (IRI) is a two-step condition: an initial imbalance of blood and oxygen supply followed by subsequent restoration of perfusion and re-oxygenation. Renal damage following IRI includes cell stress, a significant burst of free radicals, tubular damage (Acute Tubular Necrosis, ATN), endothelial activation associated with an inflammatory immune response leading to extensive cell injury, necrosis, and fibrosis [6]. The susceptibility to IRI can be attenuated by ischemic preconditioning (IPC) [7]. The IPC is defined as brief and intermittent ischemia and reperfusion episodes applied before prolonged ischemia. The degree of IPC-induced cytoprotection in the kidney depends on the duration of ischemia and reperfusion periods of each specific protocol [8, 9]. Microvascular, functional, and metabolic differences among renal cortex and medulla determine the differential regional effects of ischemia in kidney tissue [10] and may also determine the protective effects of IPC.
Both ischemia and the reperfusion phases are associated with the production of reactive oxygen species, inducing endoplasmic reticulum (ER) stress, and impairing the structure and function of renal tubular cells and glomerular cells [11-16]. Hsp27 is a stress protein that shows an early and transient increase after acute ischemia [17], inhibiting apoptosis by decreasing intracellular reactive oxygen species and the mitochondrial caspase-dependent apoptotic pathway [18]. Selective renal overexpression of Hsp27 in mice through lentiviral gene delivery protects against ischemic renal injury [19]. Cellular autophagy is activated in response to misfolded or aggregated proteins following ER stress [20], as is observed during renal IRI [21], proving a protective mechanism in the kidney for cell survival during IRI [22, 23].
Clusterin (CLU) is a chaperone-like glycoprotein [24] that promotes pro-survival autophagy [25], CLU suppresses macrophage infiltration and proinflammatory M1 during the recovery period following IRI, which may be partly responsible for tissue repair in the kidneys of WT mice after injury [26-28]. Interestingly, upregulation of CLU seems to be protective because CLU knockout (KO) mice showed increased susceptibility to renal IRI and impaired renal repair by IRI [29, 30]. However, the differential effects in renal cortex vs. medulla in Hsp27 and clusterin of IPC immediately before I/R are unknown.
Neutrophil gelatinase-associated lipocalin (NGAL) is a protein expressed by kidney tubular cells in response to kidney injury [31, 32]. Among several potential protective mechanisms at the cellular level [33-35], it has been proposed that IPC may attenuate IRI by decreasing the NF-κB DNA binding [36] and downregulating the NGAL expression [37].
Available evidence indicates that the development of vascular rarefaction and fibrosis determining potential recovery or progression to chronic renal failure is somehow dependent on the differentiation of endothelial and epithelial tubular into mesenchymal cells. These processes are called epithelial-to-mesenchymal transition (EMT) and endothelial-to-mesenchymal transition (EndMT), respectively [38, 39]. EMT is reversible, leading to the loss of cellular polarity, development of migratory capacity, resistance to apoptosis, and extracellular matrix synthesis [40-42]. EndMT resembles EMT [43, 44] and leads to the loss of cellular adhesion molecules, cytoskeletal reorganization, change of the compacted cobblestone-like into spindle-shaped phenotype without polarity, associated with reduced expression of endothelial markers (e.g., vascular endothelial-cadherin, CD31/PECAM-1, and von Willebrand Factor). Both EMT and EndMT show increased expression of mesenchymal cell markers such as vimentin, fascin, and Hsp47. Renal fibrosis is mainly associated with activation of renal resident fibroblast, which transdifferentiate into myofibroblast. However, through EMT or EndMT, a significant number of cells differentiate into fibroblasts [45-48].
Our previous study allowed us to standardize a protective IPC protocol and identify several molecular targets that were differentially modulated in the kidney cortex and medulla after I/R and IPC immediately before I/R. In the present study, we evaluated if IPC protective action implies an effect on GSH/GSSG (reduced/oxidized glutathione ratio), lipoperoxidation, and Glutathione S-Transferase activity (GST). We further characterized renal damage, oxidative stress, and the differential expression of proteostasis proteins, inflammation, and mesenchymal transition markers. The results show that the IPC protocol protected mainly the renal cortex by mechanisms dependent on Glutathione S-Transferase activity, clusterin, IL-10, and Hsp27 associated with improved proteostasis, lower inflammation, and reduced mesenchymal transition.
Materials and Methods
Animals
We used a validated model of renal ischemia and reperfusion injury using the BALB/c mice that were also used in our previous study [49]. Adult mice (20-25g) were housed in a 12h light/ dark cycle. The mice were maintained at the University de los Andes Animal Care Facility. Animals had food and water ad libitum.
Ischemia-reperfusion (I/R) and ischemia preconditioning experiments (IPC)
Animals were anesthetized with 25mg/kg i.p. ketamine /15mg/kg i.p. xylazine and maintained on a 37 °C blanket during the surgical procedure. A flank incision exposed both kidneys, and the renal pedicle was occluded for 30 min with a non-traumatic vascular clamp (cat N° 18055-02 Fine Science Tools). Renal blood flow was re-established (reperfusion stage) by clamp removal, and both incisions were sutured. The acute kidney injury is observed after 48 hours of reperfusion. According to our previous results using the murine model of IRI in BALB/c mice [49], the lower GFR values and higher values of injury and acute inflammation biomarkers are present 48 hours after reperfusion. The sham animals were subjected to the same surgery process, but they did not undergo renal pedicle occlusion [50]. In the ischemic preconditioning (IPC) protocol, animals were subjected to two cycles of 5 min of ischemia followed by 5 min of reperfusion each (IPC), immediately before the ischemia and reperfusion (I/R) [51]. Following protocols, mice were euthanized with CO2, and kidneys were dissected and processed for Western blotting, qRT-PCR, histochemistry, and biochemical analyses. During reperfusion time, the glomerular filtration rate was measured as described below.
Glomerular filtration rate (GFR) measurement
GFR was determined in conscious unrestrained mice with 48 hours of reperfusion using the excretion kinetics of an intravenous bolus of fluorescein isothiocyanate-FITC-sinistrin (FITC-sinistrin) (6 mg/100g body weight; dissolved in NaCl 0,9% Fresenius Kabi, Austria) using a miniaturized fluorometric detector (NIC-Kidney excitation 480 nm/emission 521 nm; MediBeacon, St. Lous, USA). GFR was calculated using the half-life derived from the rate constant of the single exponential phase of the FITC-sinistrin excretion curve, as previously described [52, 53].
Oxidative stress assessment
Kidney samples were homogenized in a Dounce homogenizer in 0.15M KCl. After measuring the total protein concentration [54], aliquots were deproteinized with cold trichloroacetic acid 30% V/V for 10 min, and the pellet was removed by centrifugation. Oxidative stress was determined in kidney homogenates and evaluated as changes of the GSH pool assayed in deproteinized kidney homogenates [55] and lipid peroxidation, measured as thiobarbituric acid reactive substances (TBARS) [56]. The total enzymatic GST activity was assayed in homogenates, using 1-chloro-2,4-dinitrobenzene (CDNB) as substrate and GSH as a cofactor.
Quantitative Real-Time PCR
Total RNA was isolated using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s directions. Extracted RNA was quantified at 260-nm in a NanoDrop Spectrophotometer (NanoDrop Technologies) and the integrity of the RNA was assessed by agarose gel electrophoresis. cDNA was prepared from total RNA (1μg) using a reverse transcription system (random hexamers, Improm II Reverse Transcriptase System from Promega). Then, PCR was performed duplicated for each experiment (HotStart Taq DNA polymerase from Qiagen or BRILLIANT III ULTRA-FAST SYBR GREEN QPCR (Stratagene). Amplicons were detected for Real-Time Fluorescence Detection (Rotor-Gene Q, Qiagen). The primers used are detailed in Table 1. Relative mRNA abundance was calculated using Ct values and normalized to the relative abundance of each transcript.
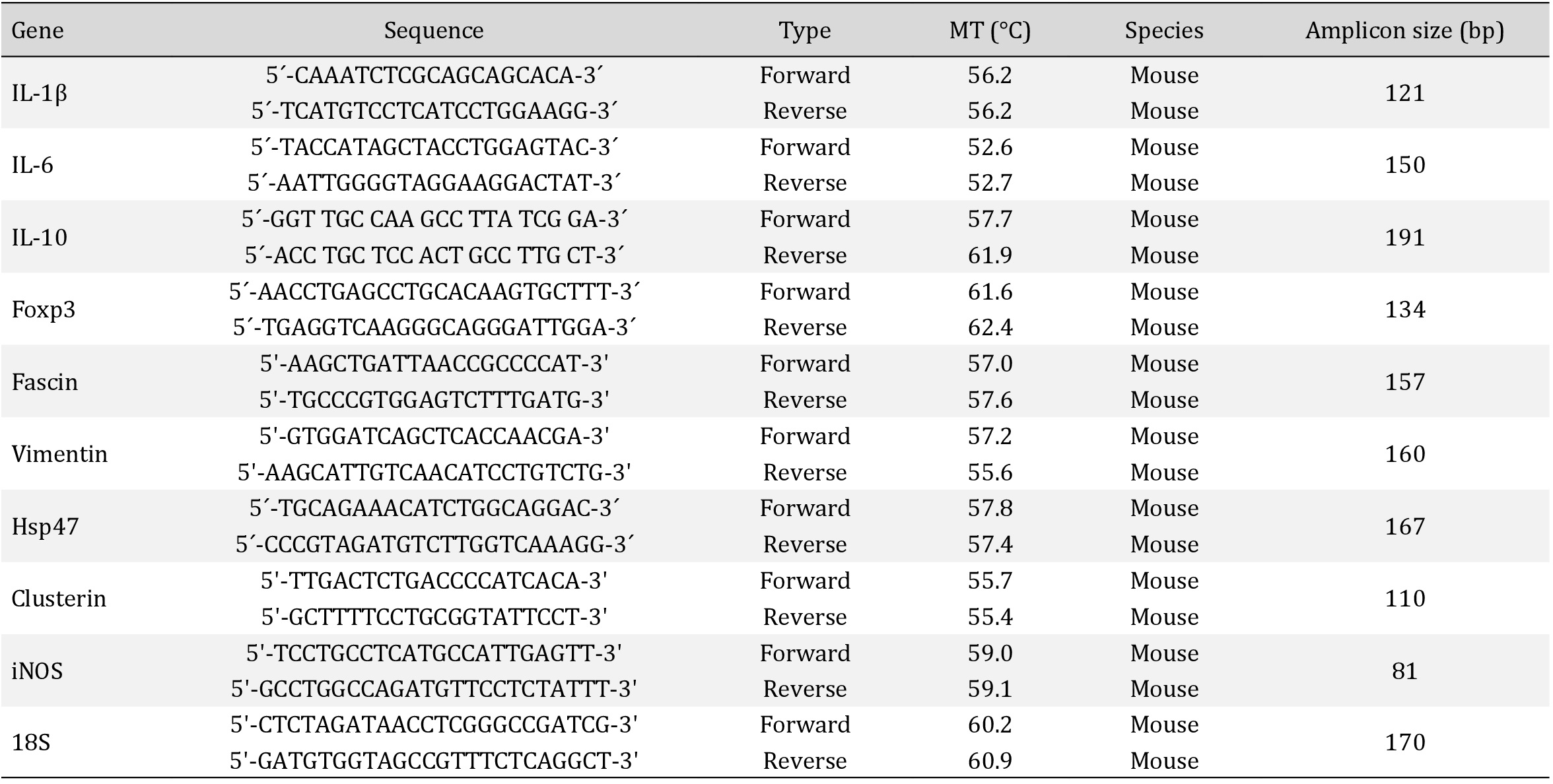
Real-time PCR primer sequences
Histochemical analysis and tissue damage determination
Kidneys were fixed in 10% formalin, included in paraffin. The kidneys were fixed in 10% buffered formalin, embedded in paraffin, sectioned, dewaxed, rehydrated, rinsed in water, and stained with hematoxylin-eosin (HE) and periodic acid Schiff (PAS). The morphologic analysis was carried out in a blinded manner as detailed previously by Dr. Luis Contreras, pathologist [49]. The cortex and medulla were evaluated for epithelial necrosis, loss of brush border, tubular dilation, and tubular congestion, among other kidney alterations observed in AKI response.
Western blot assay
Western blot was realized as was previously published with some modifications [49]. Briefly, the renal cortex and medulla were dissected and homogenized with an Ultra-Turrax homogenizer in ice-cooled 10 mM Tris·HCl buffer at pH 7.4, supplemented with 1 mM EDTA, 1 mM EGTA, 0.25 M sucrose, 1% vol/vol Triton X-100, and a protease inhibitor cocktail (Complete Mini, Roche Applied Science). Tissue homogenates were subject to centrifugation. Step one was 900×g by 10 min. Next, the tissue was sonicated for 30 seconds on ice, vortexed, and centrifugated again by 2400 × g and 17000 × by 5 min. All procedure was at 4°C. Total proteins in supernatants were measured using the BCA Protein Assay Kit (ThermoFisher Scientific), and samples were stored at -80°C. The antibodies were: rabbit anti-PECAM (77699, Cell signaling), rabbit anti-AQP-1 (ab168387-Abcam). Vimentin (clone 40E-C), and fascin (CPTC-Fascin1-3) antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development (NICHD) and maintained by the University of Iowa, Department of Biological Sciences, (Iowa City, IA). The anti-Hsp27 (sc-13132; Santa Cruz) and mouse anti-β-actin (A5441; Sigma) antibodies. Secondary antibodies were anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor-750 (Thermo Scientific). The resulting band intensities were quantified using Odyssey equipment and Image Studio Lite software (version 5.25; Li-Cor).
Statistical analysis
We use GraphPad Software to run the statistical analysis. The GFR and NGAL levels were studied using non-parametric Kruskal-Wallis analysis of variance (ANOVA). The GST activity, GSH/GSSG ratio, and thiobarbituric acid reactive substances levels were analyzed in the studied group by non-parametric Mann-Whitney analysis (t-Test). Also, the same criteria for t-Test generate the differences in the IL-1β, IL-6, Foxp3, and IL-10, AQP-1, PECAM-1, iNOS, Hsp27, clusterin, vimentin, fascin, and Hsp47 levels belong to sham, I/R, and IPC.
Results
IPC ameliorated the renal function impaired by I/R
We used a validated protocol of murine renal I/R [49]. Kidney injury was assessed through the serum NGAL (tubular damage biomarker), GFR, and kidney histology. The NGAL serum concentration was significantly upregulated (2.6-fold) in plasma samples from mice subjected to I/R compared with sham mice (111.0±22.7 for I/R and 43.3±6.3pg/mL for sham; p<0.05). The IPC protocol completely prevented the NGAL upregulation (41.8±12.0pg/mL) produced by I/R (Fig. 1A). After 48 hours of I/R, the GFR was significantly decreased by 50% of sham levels (869.2±62.0 to sham and 476.7±56.0 to I/R; µL/min/100g body weight; p<0.05). The IPC protocol significantly prevented the GFR loss observed by I/R (721.0±75.0 µL/min/100g body weight; p<0.05), reaching 83% of the sham levels (Fig. 1B). The Kidney histological analysis revealed normal kidney morphology in the cortex and medulla of the control animals. Glomeruli and tubules had normal structures (Fig. 1C sham arrows). In contrast in the I/R group, acute tubular necrosis (ATN) was present in the kidney, characterized by loss of the brush border and intratubular cellular detritus (Fig. 1C I/R, arrows). Morphological changes included other tubular epithelial alterations such as loss of the nucleus in epithelial cells and vascular congestion (Fig. 1C). In the medulla, was also observed ATN (arrows). In contrast, moderate degeneration and tubular necrosis were observed in the renal cortex from mice exposed to IPC, but in the medulla still was observed some areas of ATN and intratubular cellular detritus (arrow). Therefore, the preconditioning protocol improved the GFR and reduced the NGAL in blood but had a better histological sign of protection in the cortex than the medulla caused by the I/R.
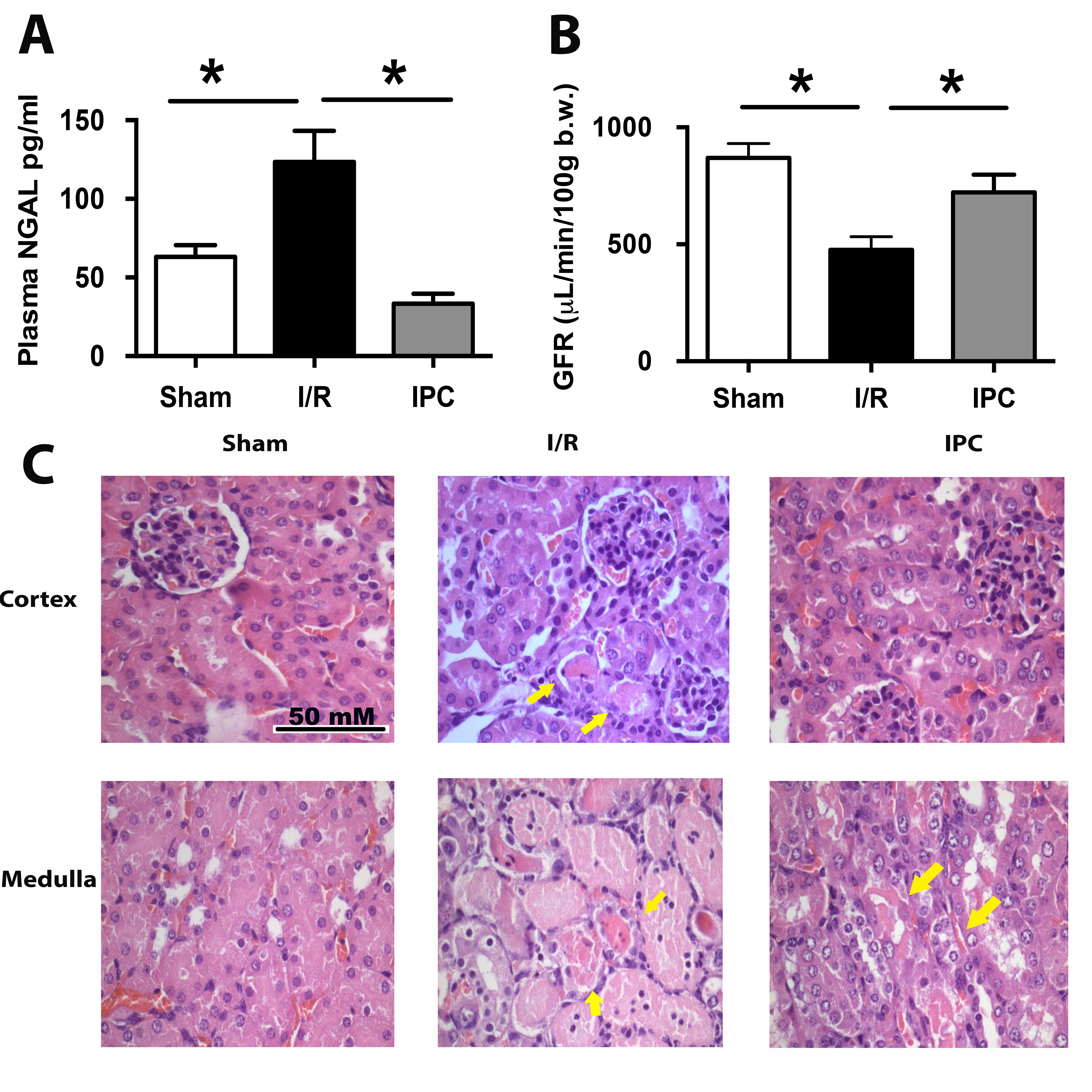
IPC protects the Epithelial and Endothelial I/R-damage in the cortex kidney
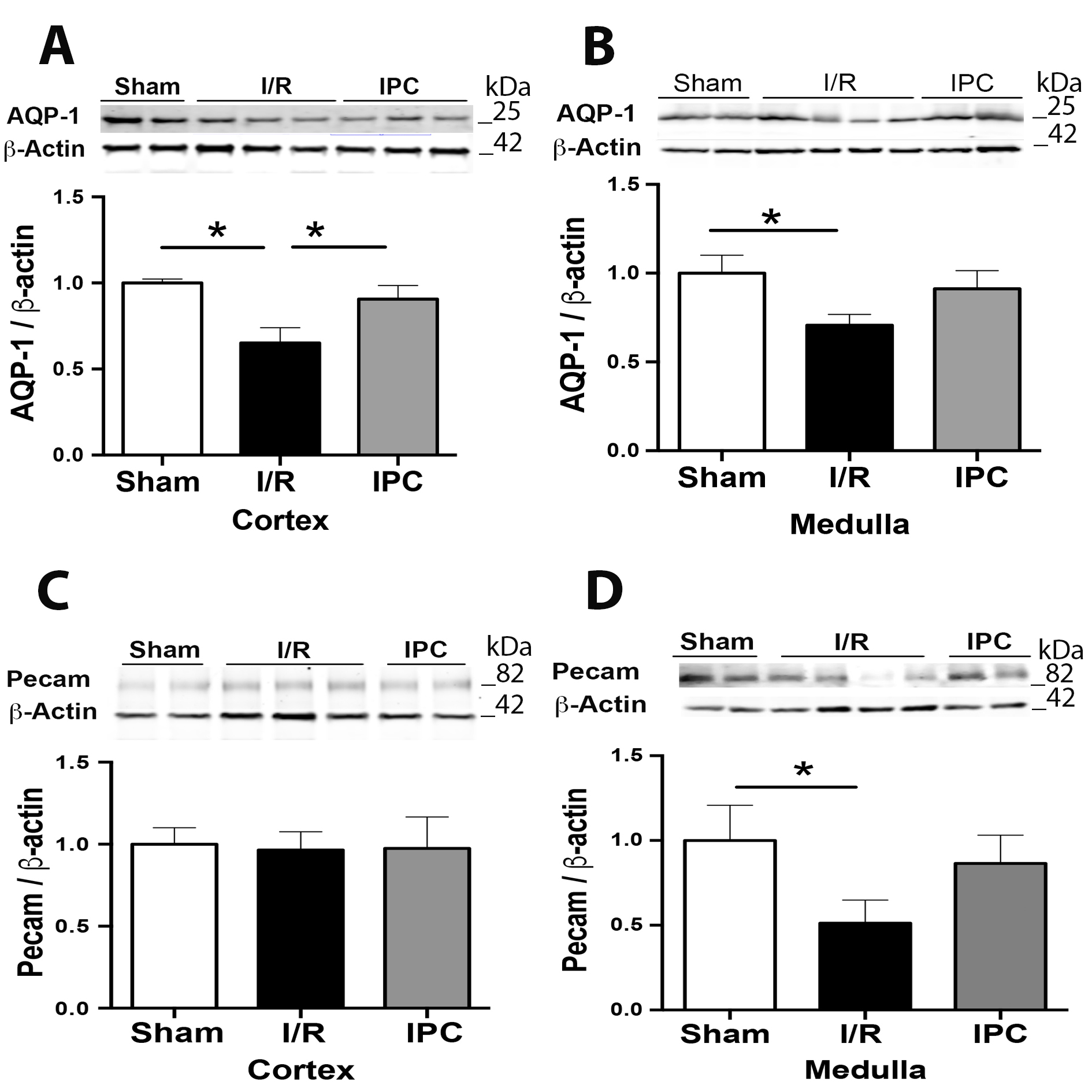
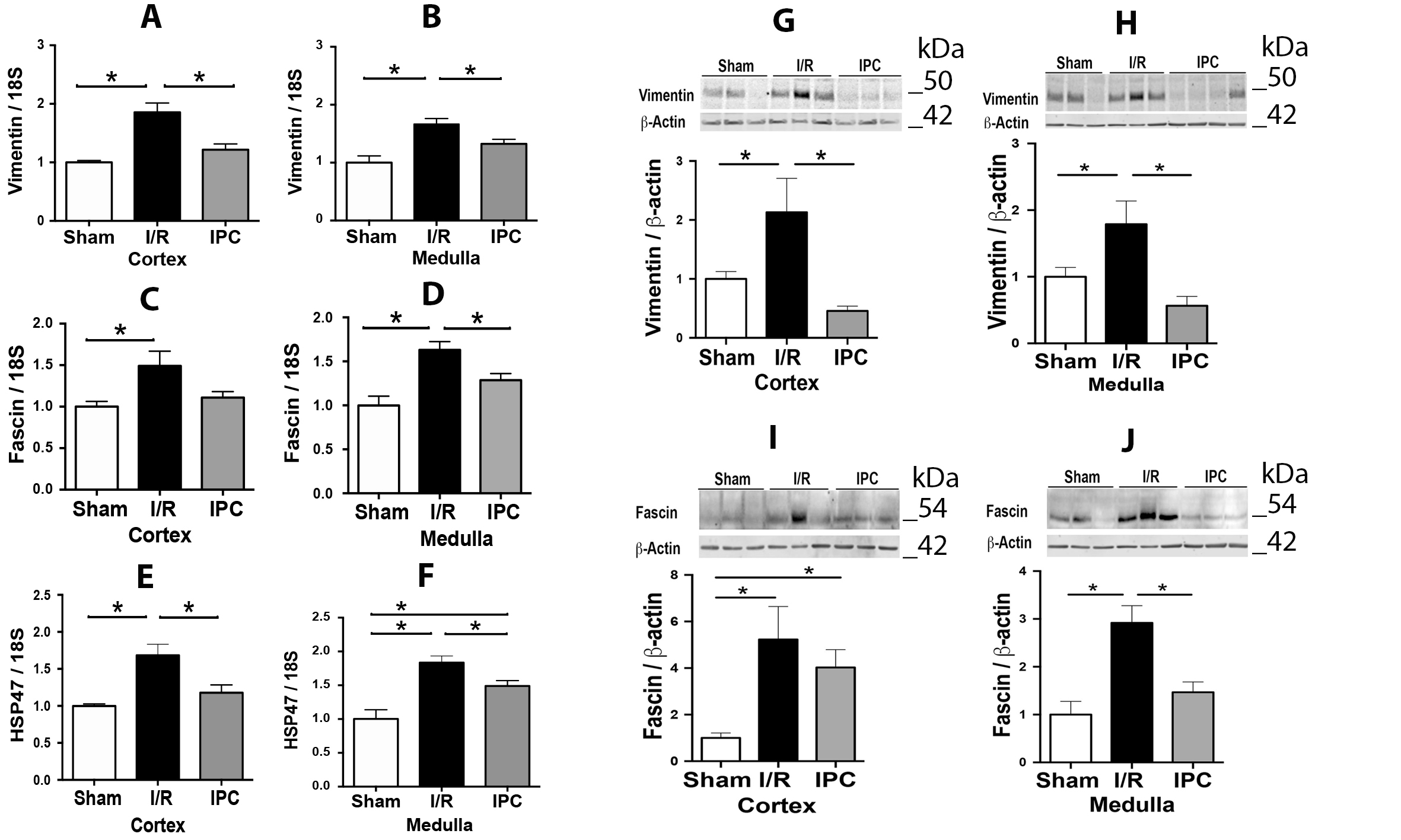
The AQP-1 is an essential protein located in the proximal and descending thin limb epithelium tubule and is implicated in the water body homeostasis [57]. We previously published the kidney AQP-1 downregulation in the cortex and medulla by renal ischemia (30 min) and reperfusion (48 hours) [49]. Here we confirmed those results (Fig. 2A-B). We found for the first time that IPC protocol significantly prevented the loss of AQP-1 protein abundance observed during I/R in the cortex, but not in the medulla. We also studied the expression of endothelial marker PECAM-1 (platelet/endothelial cell adhesion molecule 1 or CD31), a transmembrane adhesion protein expressed in kidney endothelial cells [58-60]. We found decreased PECAM-1 expression in the medulla but not in the cortex by renal I/R (Fig. 2C-D). However, the IPC did not appear to be entirely prevented the PECAM-1 downregulation (p=0.12), suggesting that IPC did not entirely protect the epithelial and endothelial cells in the medulla.
The Glutathione S-transferase activity was lost by I/R and recovered during IPC
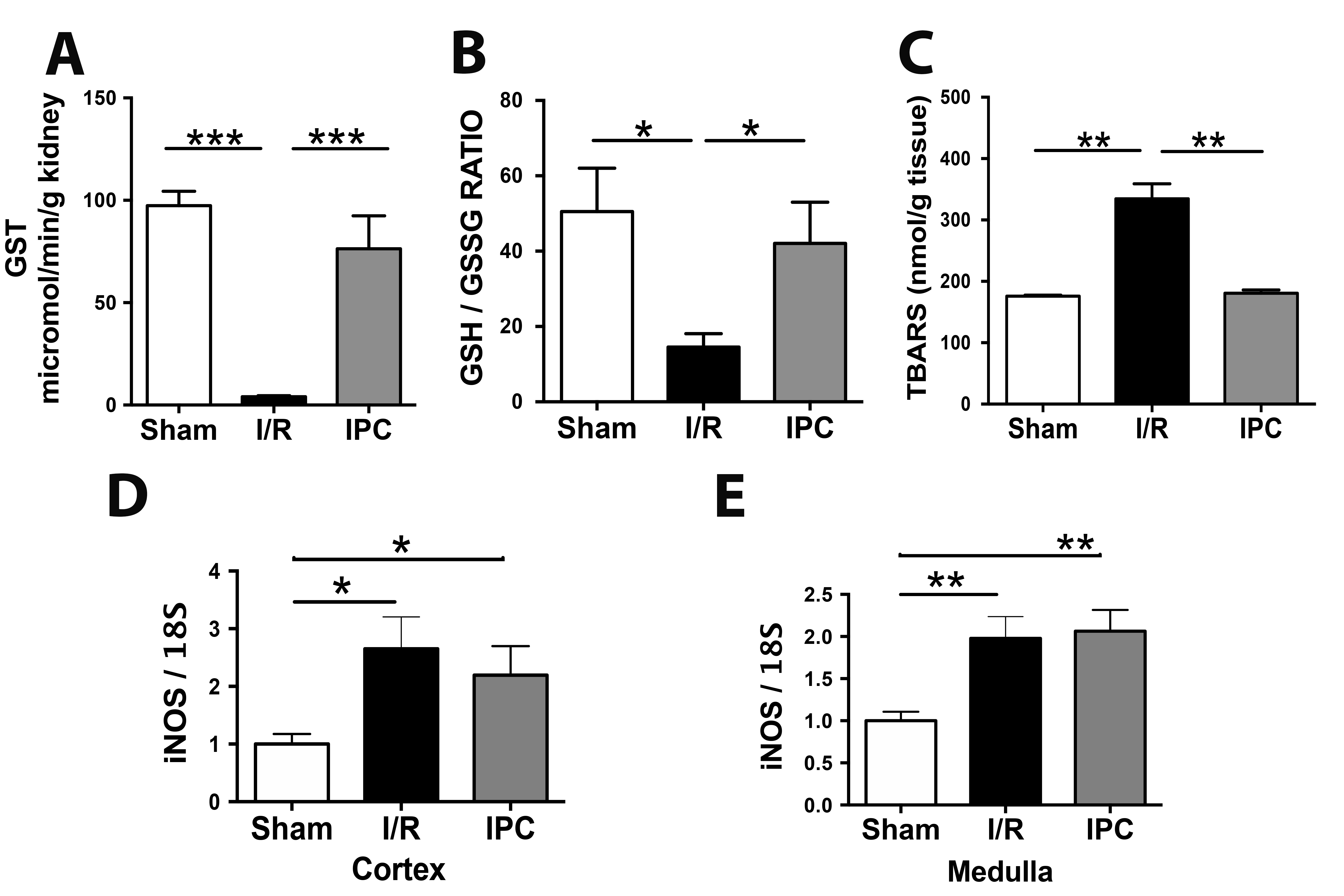
We studied the effect of renal ischemia and reperfusion injury in the oxidative stress level in the whole kidney, measuring the GSH-to-GSSG ratio to measure the soluble antioxidant status, Glutathione-S Transferase (GST) activity as an antioxidant mechanism, and TBARS as a readout for lipid peroxidation. In addition, we quantified the inducible form of NOS mRNA as an indicator of nitric oxide (NO) production. Mice subjected to renal I/R showed a significant decrease in the GSH/GSSH ratio compared to the sham group (50.48+11.55 for sham and 15.52+4.54 for I/R; nmol/g tissue, p<0.05) and IPC prevented this phenomenon (40.07+11.62 nmol/g tissue, p<0.05) (Fig. 3A). The renal lipid peroxidation (TBARS) status was increased 2 times during I/R compared to sham (175.8±1.7 for sham and 334.3±24.48 for I/R; nmol/g tissue, p<0.05). The IPC protocol reduced the TBARS at a similar level to the sham group (180.58+5.37 nmol/g tissue, p>0.05) (Fig. 3B), suggesting a strong antioxidant effect of the IPC protocol. We previously described that the GST activity was strongly reduced in the kidney by renal I/R in mice. Here, we reproduced those results (97.40+7.03 for sham and 4.37±0.48 for I/R; nmol/g tissue, p<0.05) and provided the first evidence describing the beneficial effect of IPC protocol on the GST activity (76.35+16.06 nmol/g tissue) (Fig. 3C), producing 78% of the GST activity observed in sham animals. Therefore, these data suggest that the IPC protocol prevents oxidative stress damage through an important contribution of GST activity. Interestingly, the iNOS mRNA levels increased after I/R, but the IPC did not prevent the iNOS mRNA upregulation (Fig. 3D).
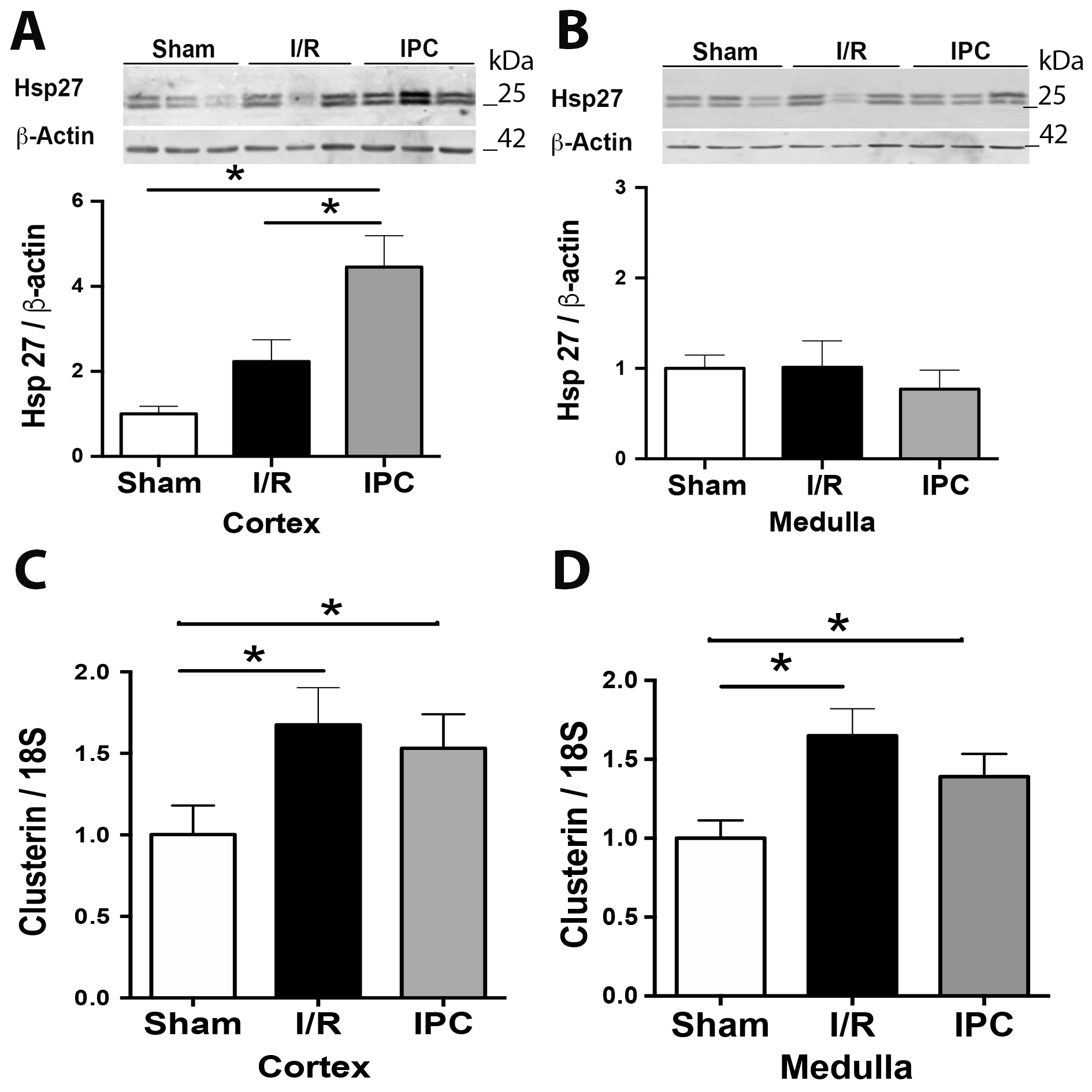
The effect of IPC in Hsp27 and Clusterin kidney expression
The Hsp27 (heat shock protein 27) is a chaperone and antioxidant protein whose induction follows injury protects against kidney injury [61]. We noted that the Hsp27 protein expression did not increase significantly by I/R in the cortex and medulla. However, the IPC treatment significantly increased the expression of Hsp27 when is compared with I/R in the cortex, but not in the medulla (Fig. 4A-B). On the other hand, we have previously reported that clusterin was increased in kidneys subjected to I/R, and it was downregulated by iNOS inhibition [49]. Here, we also showed that the mRNA level of clusterin was significantly increased in both cortex and medulla in the I/R compared with sham animals. Remarkably, the IPC protocol remains the clusterin upregulation observed by I/R (Fig. 4C-D).
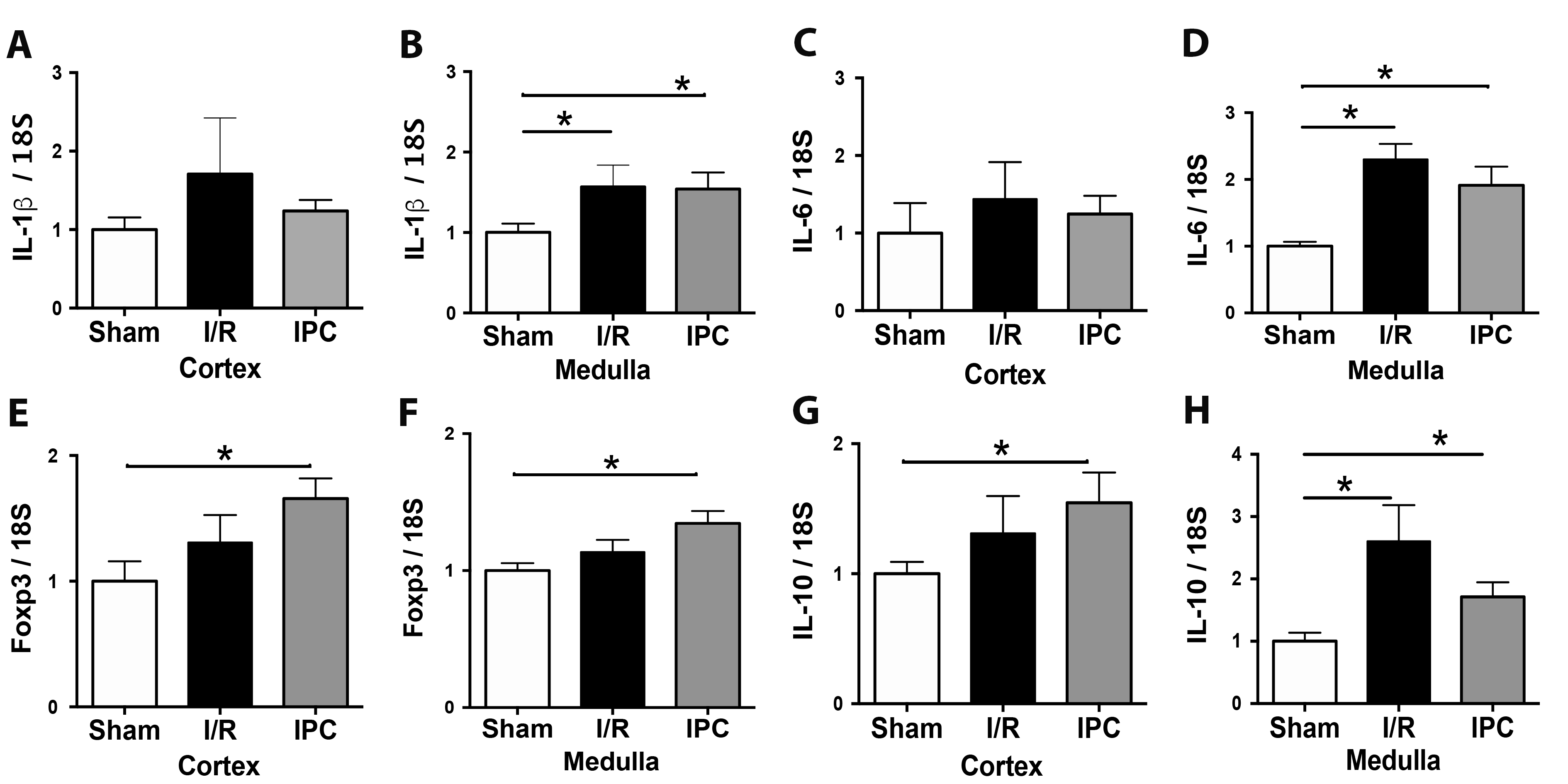
Effect of IPC in the inflammatory response
Inflammation is one of the most critical stages in renal injury after ischemia and reperfusion. We investigated the mRNA expression of two pro-inflammatory (IL-1β and IL-6) cytokines and two regulators of inflammation (Foxp3 and IL-10). In the kidney cortex, after 48 hours of I/R, we did not find changes in the mRNA levels of cytokines or Foxp3. Remarkably, the Foxp3 and IL-10 mRNAs were upregulated significantly by IPC in the cortex. In the kidney medulla, the IL-1β, IL-6, and IL-10 mRNAs increased after 48 hours of I/R, and IPC did not prevent the cytokines upregulation. In addition, IL-10 and Foxp3 mRNA were higher than the sham group in the medulla (Fig. 5B, D, F, and H).
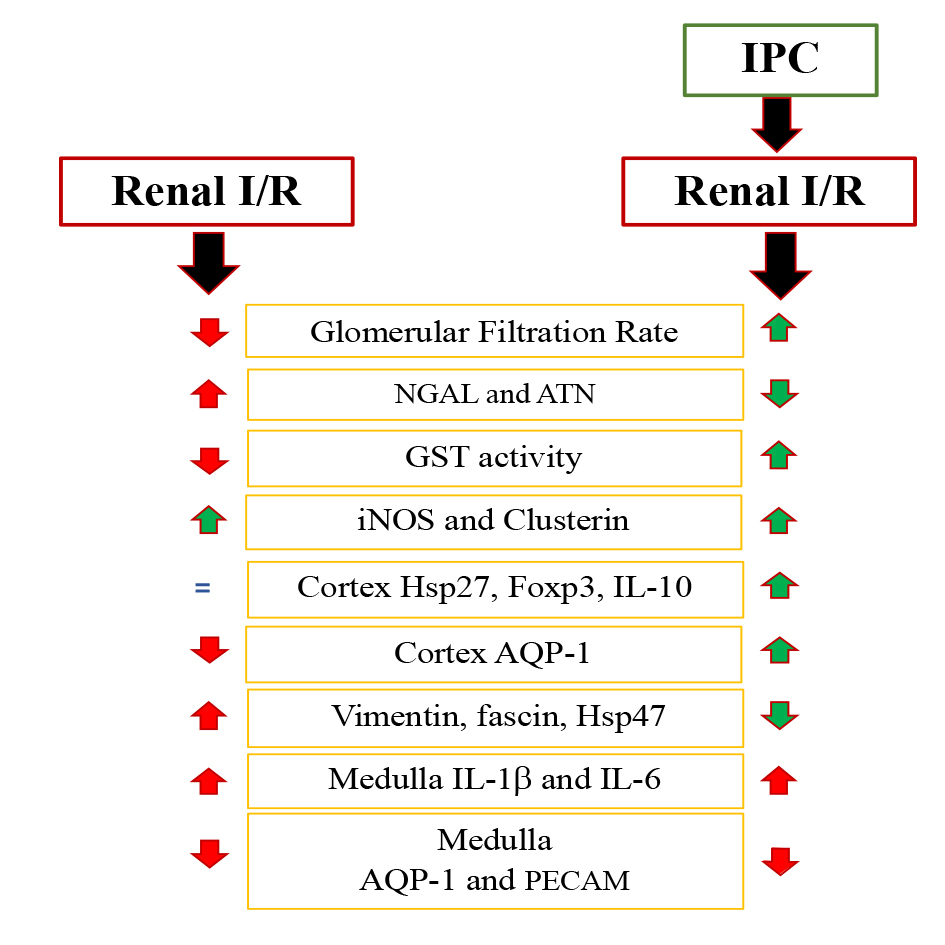
The major findings of this study are 1) IPC prevented the GFR reduction, the increase in NGAL blood levels, and reduced the signs of morphological damage principally in the cortex produced by I/R. 2) IPC improved the GST activity and prevented the decrease of GSH levels in the kidney, reducing lipoperoxidation. However, the IPC did not prevent the iNOS mRNA upregulation. 3) The epithelial and endothelial components were protected in the cortex by IPC, but not in the medulla. 4) Additional protective factors, such as Foxp3, IL-10 mRNA, and Hsp27 were upregulated preferentially in the renal cortex of IPC. 5) According to a panel of mesenchymal markers (vimentin, Fascin, and Hsp47), IPC reduced the upregulation of mesenchymal markers in the cortex and medulla compared to I/R. However, Fascin in the cortex and Hsp47 in the medulla remain higher in IPC than Sham group.
The effect of IPC on oxidative stress
One possible mechanism mediating the beneficial effects of IPC shown in the present study may imply metabolic protection. Recent studies in renal tubular cells subjected to ischemia in vitro have shown metabolic adaptation/protection from hypoxia via HIF1α due to maintenance of glycogen stores, NADH intracellular levels, and GSH//GSSG ratio [62]. Our results suggest that IPC may support the GSH levels and the GST activity.
We previously described that downregulation of GSH/GSSG ratio, GST activity, and increased lipoperoxidation observed by I/R was prevented by the pre-treatment with an iNOS inhibitor (l-NIL) [49]. In the present study, we found that IPC remains the iNOS mRNA upregulation in the same levels of I/R. Previous studies suggested a renoprotective role of iNOS during IPC. The pharmacological inhibition of iNOS or the genetic ablation of iNOS in animals resulted in the loss of the renal protective effect of IPC, indicating that iNOS activity is necessary for the development of tolerance to ischemia-induced by IPC [63]. In addition, IPC protection from cerebral ischemia was dependent on iNOS [35]. In contrast, during renal I/R, NO-derived from iNOS can dampen compensatory changes in renal hemodynamics and affect tubular reabsorption and oxygen consumption [64-67]. Thus, we speculate that the iNOS upregulation by IPC without signs of oxidative stress may explain the beneficial effects of iNOS activity during IPC.
The effect of IPC on the inflammatory response
Our results showed that IPC increased Foxp3 and IL-10 mRNA in the cortex without upregulation of IL-1β or IL-6 mRNA, suggesting that this condition may be one mechanism that confers protection to the renal cortex by IPC. These results suggest that a potential mechanism mediating the protective effect of IPC is the polarization/recruitment of Treg lymphocytes, which may ameliorate and/or limit the inflammatory response triggered by I/R. Previous studies indicated that IL-10 protects the kidney after renal ischemia injury by suppressing inflammatory mediators such as TNF-α and IL-6 [68, 69] and inhibiting leukocyte adhesion on the endothelium [70]. Compared to wild-type (WT) mice, IL-10 knockout mice with IRI showed a more marked reduction of renal function, upregulated expression of AKI biomarkers, increased expression of the pro-inflammatory cytokines, and increased expression of pro-apoptosis factors [71]. We did not observe significant changes in IL-1β or IL-6 mRNA abundance in the renal cortex after 48 hours of reperfusion. However, future studies are necessary to clarify if proinflammatory cytokines such as IL-6 presented an early increase after I/R in the renal cortex, which may have been ameliorated by IL-10 upregulation after preconditioning. Indeed, the study of Sasaki et al., showed a transient increase in renal IL-6 mRNA levels at 5- and 24-hours post-I/R, which returned at 48 hours to levels similar to those observed in sham surgery mice.
The effect of IPC on endothelial and epithelial cell alterations
Renal injury following ischemia and reperfusion provokes disruption of microvasculature due to endothelial cell damage and renal dysfunction following the loss of epithelial cell polarity secondary to cytoskeletal alterations. AQP-1 is a protein expressed at the apical and basolateral region of the proximal tubule epithelial cells and the thin descending limb of Henle and in microvascular endothelia, including vasa recta [72]. Our study shows a differential effect of IPC in the cortex than medulla. We and others described previously that I/R decreases the expression of AQP-1 in the renal cortex and medulla. Here we observed that AQP-1 lower expression after I/R was prevented by IPC in the cortex; only a non-significant tendency to protection was observed in the medulla. PECAM-1 was reduced after 48 hours of I/R in renal medulla only, and IPC did not confer protection. In contrast, vimentin, fascin, and Hsp47 were increased by I/R, and IPC reduced this upregulation in both renal regions. However, Hsp47 in medulla during IPC was kept higher than sham animals, suggesting an association between high Hsp47 and low AQP1 or PECAM-1 expression during IPC in the medulla.
The effect of IPC on Hsp27 and Clusterin
Hsp27 is a chaperone that confers cytoprotection from ischemia [73, 74] by stabilizing the actin cytoskeleton, displaying an anti-apoptotic function, reducing ATP depletion, and reactive oxygen species [75]. Hsp27 overexpression protects the heart and brain from ischemic injury [76], and recent studies show that Hsp27 is necessary for protection against renal IRI conferred by pharmacological A1 adenosine receptor (AR) activation before renal ischemia [77]. The upregulation of Hsp27 by renal injection of adenovirus encoding Hsp27 or by the injection of LPS before renal IRI was also protective, ameliorating the increase in creatinine/BUN and apoptosis up to 24 hours after I/R [78, 79]. Furthermore, Jo et al., in a delayed model of ischemic preconditioning and uninephrectomy (four cycles of 5/5 min of I/R applied for 24h before 30 min of renal ischemia in the left kidney), found increased expression of Hsp27, which was associated with the reduction of cellular injury [63]. In addition, the induction of Hsp27 before I/R prevented the increase in hydrogen peroxide (HPO) generation up to 24 hours post-I/R, and the knockdown of Hsp27 did not cause a further increase in HPO generation after I/R as compared with control mice with unaltered basal Hsp27 expression [78]. In the present study, we found that I/R did not significantly increase the expression of Hsp27 in the cortex. However, the IPC protocol increased the Hsp27 protein expression in the renal cortex. Thus, the induction of Hsp27 in the cortex would be implicated in preventing oxidative stress obtained by IPC and probably through GST. It is tempting to speculate that similar to LPS injection, the upregulation of Hsp27 induced by IPC could be a consequence of cortex-specific activation of Hypoxia Inducible Factor 2a (HIF-2a) pathway [78]. However, LPS-stimulated renoprotection was also dependent on the upregulation of eNOS and iNOS. Here we found the upregulation of iNOS and Hsp27 in the cortex by IPC but not in the medulla making necessary further studies to clarify if the induction of Hsp27 due to IPC is somehow dependent on NO bioavailability.
Proteins enter the ER and exit as either folded or misfolded proteins. Altered redox signaling and increased reactive oxygen species (ROS) disrupt ER function and proteostasis [80]. GSH plays a core role in maintaining ER oxidoreductases in a reduced state to catalyze reduction or isomerization reactions, and under oxidative stress conditions, GSH acts as a redox buffer source, in addition to its role in disulfide bond formation. Our results showed that the IPC protocol prevented the decrease in GSH/GSSG balance and increasing the GST activity. Thus, the IPC protocol suggests help to maintain ER function and proteostasis.
Noteworthy, in agreement with other groups, we found that clusterin is induced following renal I/R [49]. however, clusterin induction was not modified by IPC, suggesting a potential protective role during IPC.
Finally, we observed differences in the effect of IRI and IPC in the cortex and medulla. In the cortex, IRI downregulated (AQP-1) and upregulated (iNOS, clusterin, vimentin, fascin1, and Hsp47) different molecular panel than medulla. Thus, IRI induced downregulation of AQP-1 and PECAM-1, but increased the IL-1β, IL-6, IL-10, iNOS, clusterin, vimentin, fascin, and Hsp47 expression in medulla. So, the results indicate that I/R has a more marked/persistent inflammatory effect in the medulla than in the cortex. Consistent with differential injury mechanisms in the cortex and medulla, IPC showed differential protective effects. IPC upregulated Hsp27 in the cortex but not in the medulla, and the markers of Treg lymphocytes, FoxP3 mRNA, and IL-10 mRNA, were also induced by IPC in the kidney cortex. The potential mechanisms that may potentiate Treg activity response in the renal cortex after IPC may depend on amelioration of injury due to enhanced tissular tolerance to transient hypoxia and lower cellular damage after I/R.
Conclusion
In summary, we observed that IPC reduced oxidative stress and tubular damage, concomitant with the upregulation of GST activity, clusterin, Foxp3, IL-10, and Hsp27 (factors involved in cell protection) evident in the cortex. Interestingly, the increase of IL-1β and IL-6 mRNA levels and the downregulation of PECAM-1 and AQP-1 protein expression in kidney medulla during IPC shows that the IPC has a preferential better effect in the cortex (Fig. 7).
In addition, during IPC the iNOS upregulation of oxidative stress, suggests a potential explication of why iNOS is good during IPC and bad during I/R. Our results provide relevant and novel evidence to understand the molecular components involved in IPC, showing that molecular mechanisms that lead to I/R protection may be cortex and medulla specific. In addition, we described new components associated with IPC, such as clusterin and GST, that are potential targets of future investigation.
Author Contributions
Conceived and designed experiments: CEI.
Performed the experiments: CP, YH; ML, JR, CA, JL, LM.
Analyzed the data: CP, ML, JL, LM, CEI.
Wrote the paper CP, LM, CEI.
Contributed reagents/materials/analysis tools: CEI, LM, CA. JL, CP.
Funding Sources
The present study was financially supported through grants from the FONDECYT-1151157, FONDECYT-21150304, FAI (Carlos E. Irarrázabal), and FAI-Puente (Consuelo Pasten).
Statement of Ethics
All experimental procedures were under institutional and international standards for humane care and laboratory animal use (Animal Welfare Assurance Publication A5427-01,
Office for Protection from Research Risks, Division of Animal Welfare. NIH, USA). All procedures were approved by the Committee on the Ethics of Animal Experiments of the University de los Andes, Chile.
The authors have no conflicts of interest to declare.
| 1 Bellomo R, Kellum JA, Ronco C: Acute kidney injury. Lancet 2012;380:756-766. https://doi.org/10.1016/S0140-6736(11)61454-2 |
||||
| 2 Franzin R, Stasi A, Fiorentino M, Simone S, Oberbauer R, Castellano G, Gesualdo L: Renal Delivery of Pharmacologic Agents During Machine Perfusion to Prevent Ischaemia-Reperfusion Injury: From Murine Model to Clinical Trials. Front Immunol 2021;12:673562. https://doi.org/10.3389/fimmu.2021.673562 |
||||
| 3 Pefanis A, Ierino FL, Murphy JM, Cowan PJ: Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int 2019;96:291-301. https://doi.org/10.1016/j.kint.2019.02.009 |
||||
| 4 Mehta RL: Outcomes research in acute renal failure. Semin Nephrol 2003;23:283-294. https://doi.org/10.1016/S0270-9295(03)00064-0 |
||||
| 5 Boltansky A, Bassa C, Melani S, Sepulveda A, Maldonado I, Postigo J, Sotta E, Vidueira P, Cavagnaro C, Cavada G, Benavente C, Villamizar G, Vukusich A, Irarrázabal CE: Incidence and consequences of acute kidney injury among patients admitted to critical care units. Rev Med Chil 2015;143:1114-1120. https://doi.org/10.4067/S0034-98872015000900003 |
||||
| 6 Tanaka S, Tanaka T, Nangaku M: Hypoxia as a key player in the AKI-to-CKD transition. Am J Physiol Renal Physiol 2014;307:F1187-F1195. https://doi.org/10.1152/ajprenal.00425.2014 |
||||
| 7 Torosyan R, Huang S, Bommi PV, Tiwari R, An SY, Schonfeld M, Rajendran G, Kavanaugh MA, Gibbs B, Truax AD, Bohney S, Calcutt MW, Kerr EW, Leonardi R, Gao P, Chandel NS, Kapitsinou PP: Hypoxic preconditioning protects against ischemic kidney injury through the IDO1/kynurenine pathway. Cell Rep 2021; 36:109547. https://doi.org/10.1016/j.celrep.2021.109547 |
||||
| 8 Sedaghat Z, Kadkhodaee M, Seifi B, Salehi E: Inducible and endothelial nitric oxide synthase distribution and expression with hind limb per-conditioning of the rat kidney. Arch Med Sci 2019;15:1081-1091. https://doi.org/10.5114/aoms.2019.85651 |
||||
| 9 Choi HS, Hwang JK, Kim JG, Hwang HS, Lee SJ, Chang YK, Kim JI, Moon IS: The optimal duration of ischemic preconditioning for renal ischemia-reperfusion injury in mice. Ann Surg Treat Res 2017;93:209-216. https://doi.org/10.4174/astr.2017.93.4.209 |
||||
| 10 Ray SC, Mason J, O'Connor PM: Ischemic Renal Injury: Can Renal Anatomy and Associated Vascular Congestion Explain Why the Medulla and Not the Cortex Is Where the Trouble Starts? Semin Nephrol 2019;39:520-529. https://doi.org/10.1016/j.semnephrol.2019.10.002 |
||||
| 11 Delgado-Valero B, de la Fuente-Chávez L, Romero-Miranda A, Visitación Bartolomé M, Ramchandani B, Islas F, Luaces M, Cachofeiro V, Martínez-Martínez E: Role of endoplasmic reticulum stress in renal damage after myocardial infarction. Clin Sci (Lond) 2021;135:143-159. https://doi.org/10.1042/CS20201137 |
||||
| 12 Sareen-Khanna K, Papillon J, Wing SS, Cybulsky AV: Role of the deubiquitinating enzyme ubiquitin-specific protease-14 in proteostasis in renal cells. Am J Physiol Renal Physiol 2016;311:F1035-F1046. https://doi.org/10.1152/ajprenal.00252.2016 |
||||
| 13 Oliva, J: Proteasome and organs ischemia-reperfusion injury. Int J Mol Sci 2017;19:E106. https://doi.org/10.3390/ijms19010106 |
||||
| 14 Cybulsky AV, Takano T, Papillon J, Bijian K: Role of the endoplasmic reticulum unfolded protein response in glomerular epithelial cell injury. J Biol Chem 2005;280:24396-24403, https://doi.org/10.1074/jbc.M500729200 |
||||
| 15 Gu Y, Huang F, Wang Y, Chen C, Wu S, Zhou S, Hei Z, Yuan D: Connexin32 plays a crucial role in ROS-mediated endoplasmic reticulum stress apoptosis signaling pathway in ischemia reperfusion-induced acute kidney injury. J Transl Med 2018;16:117. https://doi.org/10.1186/s12967-018-1493-8 |
||||
| 16 Uddin MJ, Jeong J, Pak ES, Ha H: CO-Releasing Molecule-2 Prevents Acute Kidney Injury through Suppression of ROS-Fyn-ER Stress Signaling in Mouse Model. Oxid Med Cell Longev 2021;2021:9947772. https://doi.org/10.1155/2021/9947772 |
||||
| 17 Guo Q, Du X, Zhao Y, Zhang D, Yue L, Wang Z: Ischemic postconditioning prevents renal ischemia reperfusion injury through the induction of heat shock proteins in rats. Mol Med Rep 2014;10:2875-2881. https://doi.org/10.3892/mmr.2014.2641 |
||||
| 18 Chebotareva N, Bobkova I, Shilov E: Heat shock proteins and kidney disease: perspectives of HSP therapy. Cell Stress Chaperones 2017;22:319-343. https://doi.org/10.1007/s12192-017-0790-0 |
||||
| 19 Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D'Agati VD, Lee HT: Selective renal overexpression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 2010;299:347-358. https://doi.org/10.1152/ajprenal.00194.2010 |
||||
| 20 Murrow L, Debnath J: Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 2013;8:105-137. https://doi.org/10.1146/annurev-pathol-020712-163918 |
||||
| 21 Jiang M, Liu K, Luo J, Dong Z: Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 2010;176:1181-1192. https://doi.org/10.2353/ajpath.2010.090594 |
||||
| 22 Isaka Y, Kimura T, Takabatake Y: The protective role of autophagy against aging and acute ischemic injury in kidney proximal tubular cells. Autophagy 2011;9:1085-1087. https://doi.org/10.4161/auto.7.9.16465 |
||||
| 23 Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T Rakugi H, Isaka Y: Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011;22:902-913. https://doi.org/10.1681/ASN.2010070705 |
||||
| 24 Satapathy S, Wilson MR: The Dual Roles of Clusterin in Extracellular and Intracellular Proteostasis. Trends Biochem 2021;46:P652-P660. https://doi.org/10.1016/j.tibs.2021.01.005 |
||||
| 25 Alnasser HA, Guan Q, Zhang F, Gleave ME, Nguan CY, Du C: Requirement of clusterin expression for prosurvival autophagy in hypoxic kidney tubular epithelial cells. Am J Physiol Renal Physiol 2016;310:F160-F173. https://doi.org/10.1152/ajprenal.00304.2015 |
||||
| 26 Weng X, Zhao H, Guan Q, Shi G, Feng S, Gleave ME, Nguan CC, Du C: Clusterin regulates macrophage expansion, polarization and phagocytic activity in response to inflammation in the kidneys. Immunol Cell Biol 2021;99:274-287. https://doi.org/10.1111/imcb.12405 |
||||
| 27 Sarkar S, Sinha R, Chaudhury AR, Maduwage K, Abeyagunawardena A, Bose N, Pradhan S, Bresolin NL, Garcia BA, McCulloch M: Snake bite associated with acute kidney injury. Pediatr Nephrol 2021; DOI: 10.1007/s00467-020-94911-x. https://doi.org/10.1007/s00467-020-04911-x |
||||
| 28 Guo J, Guan Q, Liu X, Wang H, Gleave ME, Nguan CY, Du C: Relationship of clusterin with renal inflammation and fibrosis after the recovery phase of ischemia-reperfusion injury. BMC Nephrol 2016;17:133. https://doi.org/10.1186/s12882-016-0348-x |
||||
| 29 Nguan CY, Guan Q, Gleave ME, Du C: Promotion of cell proliferation by clusterin in the renal tissue repair phase after ischemia-reperfusion injury. Am J Physiol Renal Physiol 2014;306:F724-F733. https://doi.org/10.1152/ajprenal.00410.2013 |
||||
| 30 Zhou W, Guan Q, Kwan CC, Chen H, Gleave ME, Nguan CY, Du C: Loss of clusterin expression worsens renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 2010;298:F568-F578. https://doi.org/10.1152/ajprenal.00399.2009 |
||||
| 31 Griffin BR, Gist KM, Faubel S: Current Status of Novel Biomarkers for the Diagnosis of Acute Kidney Injury: A Historical Perspective. J Intensive Care Med. 2020;35:415-424. https://doi.org/10.1177/0885066618824531 |
||||
| 32 Ramirez-Sandoval JC, Herrington W, Morales-Buenrostro LE: Neutrophil gelatinase-associated lipocalin in kidney transplantation: A review. Transplant Rev (Orlando). 2015;29:139-144. https://doi.org/10.1016/j.trre.2015.04.004 |
||||
| 33 Ge YZ, Wu R, Xin H, Liu H, Lu TZ, Zhao YC, Shen JW, Hu ZK, Yu P, Zhou LH, Xu LW, Xu Z, Wu JP, Li WC, Zhu JG, Jia RP: Effects of ischemic preconditioning on the systemic and renal hemodynamic changes in renal ischemia reperfusion injury. Int J Clin Exp Pathol 2015;8:1128-1140. | ||||
| 34 Chen X, Liu X, Wan X, Wu Y, Chen Y, Cao C: Ischemic preconditioning attenuates renal ischemia-reperfusion injury by inhibiting activation of IKKβ and inflammatory response. Am J Nephrol 2009;30:287-294. https://doi.org/10.1159/000225928 |
||||
| 35 Cho S, Park E-M, Zhou P, Frys K, Ross ME, Iadecola C: Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab 2005;25:493-501. https://doi.org/10.1038/sj.jcbfm.9600058 |
||||
| 36 Li JR, Ou YC, Wu CC, Wang JD, Lin SY, Wang YY, Chen WY, Chen CJ: Ischemic preconditioning improved renal ischemia/reperfusion injury and hyperglycemia. IUBMB Life 2019;71:321-329. https://doi.org/10.1002/iub.1972 |
||||
| 37 Xie Y, Xiao J, Fu C, Zhang Z, Ye Z, Zhang X: Ischemic Preconditioning Promotes Autophagy and Alleviates Renal Ischemia/Reperfusion Injury. Biomed Res Int 2018;2018:8353987. https://doi.org/10.1155/2018/8353987 |
||||
| 38 Cruz-Solbes AS, Youker K: Epithelial to Mesenchymal Transition (EMT) and Endothelial to Mesenchymal Transition (EndMT): Role and Implications in Kidney Fibrosis. Results Probl Cell Differ 2017;60:345-372. https://doi.org/10.1007/978-3-319-51436-9_13 |
||||
| 39 Lamouille S, Xu J, Derynck R: Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178-196. https://doi.org/10.1038/nrm3758 |
||||
| 40 Sheng L, Zhuang S: New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front Physiol 2020;11:569322. https://doi.org/10.3389/fphys.2020.569322 |
||||
| 41 Lim J, Thiery JP: Epithelial-mesenchymal transitions: insights from development. Development 2012;139:3471-3486. https://doi.org/10.1242/dev.071209 |
||||
| 42 Yamashita N, Kusaba T, Nakata T, Tomita A, Ida T, Watanabe-Uehara N, Ikeda K, Kitani T, Uehara M, Kirita Y, Matoba S, Humphreys BC, Tamagaki K: Intratubular epithelial-mesenchymal transition and tubular atrophy after kidney injury in mice. Am J Physiol Renal Physiol 2020;319:F579-F591. https://doi.org/10.1152/ajprenal.00108.2020 |
||||
| 43 Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M: Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015;125:4514-4528. https://doi.org/10.1172/JCI82719 |
||||
| 44 Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V: Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition from cardiovascular development to disease. Circulation 2012;125:1795-1808. https://doi.org/10.1161/CIRCULATIONAHA.111.040352 |
||||
| 45 Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F: Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015;131:1006-1018. https://doi.org/10.1161/CIRCULATIONAHA.114.008750 |
||||
| 46 Sato Y, Yanagita M: Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm Regen 2017;37:17. https://doi.org/10.1186/s41232-017-0048-3 |
||||
| 47 LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R: Origin and function of myofibroblasts in kidney fibrosis. Nat Med 2013;19:1047-1053. https://doi.org/10.1038/nm.3218 |
||||
| 48 Xu-Dubois YC, Ahmadpoor P, Brocheriou I, Louis K, Arzouk Snanoudj N, Rouvier P, Taupin JL, Corchia A, Galichon P, Barrou B, Giraud S, Hauet T, Jouanneau C, Rodenas A, Placier S, Niasse A, Ouchelouche S, Naimi BY, Akil E, Hertig A, et al.: Microvasculature partial endothelial mesenchymal transition in early posttransplant biopsy with acute tubular necrosis identifies poor recovery renal allografts. Am J Transplant 2020;20:2400-2412. https://doi.org/10.1111/ajt.15847 |
||||
| 49 Pasten C, Alvarado C, Rocco J, Contreras L, Aracena P, Liberona J, Suazo C, Michea L, Irarrazabal CE: l-NIL prevents the ischemia and reperfusion injury involving TLR-4, GST, clusterin, and NFAT-5 in mice. Am J Physiol Renal Physiol 2019;316:F624-F634. https://doi.org/10.1152/ajprenal.00398.2018 |
||||
| 50 Wei Q, Dong Z: Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol Renal Physiol 2012;303:F1487-94. https://doi.org/10.1152/ajprenal.00352.2012 |
||||
| 51 Mahfoudh-Boussaid A, Zaouali MA, Hadj-Ayed K, Miled AH, Saidane-Mosbahi D, Rosello-Catafau J, Ben Abdennebi H: Ischemic preconditioning reduces endoplasmic reticulum stress and upregulates hypoxia inducible factor-1α in ischemic kidney: the role of nitric oxide. J Biomed Sci 2012;19:7. https://doi.org/10.1186/1423-0127-19-71 |
||||
| 52 Giani JF, Bernstein KE, Janjulia T, Han J, Toblli JE, Shen XZ, Rodriguez-Iturbe B, McDonough AA, Gonzalez-Villalobos RA: Salt Sensitivity in Response to Renal Injury Requires Renal Angiotensin-Converting Enzyme. Hypertension 2015;66:534. https://doi.org/10.1161/HYPERTENSIONAHA.115.05320 |
||||
| 53 Schreiber A, Shulhevich Y, Geraci S, Hesser J, Stsepankou D, Neudecker S, Koenig S, Heinrich R, Hoecklin F, Pill J, Friedemann J, Schweda F, Gretz N, Schock-Kusch D: Transcutaneous measurement of renal function in conscious mice. Am J Physiol Physiol 2012;303:F783-F788. https://doi.org/10.1152/ajprenal.00279.2012 |
||||
| 54 Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951;193:265. https://doi.org/10.1016/S0021-9258(19)52451-6 |
||||
| 55 Ohkawa H, Ohishi N, Yagi K: Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979;95:351. https://doi.org/10.1016/0003-2697(79)90738-3 |
||||
| 56 Tietze F: Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 1969;27:502-522. https://doi.org/10.1016/0003-2697(69)90064-5 |
||||
| 57 Hua Y, Ying X, Qian Y, Liu H, Lan Y, Xie A, Zhu X: Physiological and pathological impact of AQP1 knockout in mice. Biosci Rep 2019;39:BSR20182303. https://doi.org/10.1042/BSR20182303 |
||||
| 58 Uil M, Butter LM, Claessen N, Larsen PW, Florquin S, Roelofs JJTH: Platelet inhibition by ticagrelor is protective against diabetic nephropathy in mice. FASEB J 2020;34:13750-13761. https://doi.org/10.1096/fj.202000897R |
||||
| 59 Kondo S, Scheef EA, Sheibani N, Sorenson CM: PECAM-1 isoform-specific regulation of kidney endothelial cell migration and capillary morphogenesis. Am J Physiol Cell Physiol 2007;292:C2070-C2083. https://doi.org/10.1152/ajpcell.00489.2006 |
||||
| 60 Dursun I, Yel S, Unsur E: Dynamics of circulating microparticles in chronic kidney disease and transplantation: Is it really reliable marker? World J Transplant 2015;5:267-275. https://doi.org/10.5500/wjt.v5.i4.267 |
||||
| 61 Vidyasagar A, Reese S, Acun Z, Hullett D, Djamali A: HSP27 is involved in the pathogenesis of kidney tubulointerstitial fibrosis. Am J Physiol Renal Physiol 2008;295:F707-F716. https://doi.org/10.1152/ajprenal.90240.2008 |
||||
| 62 Ito M, Tanaka T, Ishii T, Wakashima T, Fukui K, Nangaku M: Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int 2020;97:687-701. https://doi.org/10.1016/j.kint.2019.10.020 |
||||
| 63 Joo JD, Kim M, D'Agati VD, Lee HT: Ischemic preconditioning provides both acute and delayed protection against renal ischemia and reperfusion injury in mice. J Am Soc Nephrol 2006;17:3115-3123. https://doi.org/10.1681/ASN.2006050424 |
||||
| 64 Fatemikia H, Ketabchi F, Karimi Z, Moosavi SM: Distant effects of unilateral renal ischemia/reperfusion on contralateral kidney but not lung in rats: the roles of ROS and iNOS. Can J Physiol Pharmacol 2016;94:477-487. https://doi.org/10.1139/cjpp-2015-0285 |
||||
| 65 Park SW, Chen SW, Kim M, D'Agati VD, Lee HT: Human heat shock protein 27-overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am J Physiol Renal Physiol 2009;297:F885-F894. https://doi.org/10.1152/ajprenal.00317.2009 |
||||
| 66 Kalogeris T, Baines CP, Krenz M, Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012;298:229-317. https://doi.org/10.1016/B978-0-12-394309-5.00006-7 |
||||
| 67 Ahmad A, Dempsey SK, Daneva Z, Azam M, Li N, Li PL, Ritter JK: Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int J Mol Sci 2018;19:2605. https://doi.org/10.3390/ijms19092605 |
||||
| 68 Wan X, Huang WJ, Chen W, Xie HG, Wei P, Chen X, Cao CC: IL-10 deficiency increases renal ischemia-reperfusion injury. Nephron Exp Nephrol 2014;128:37-45. https://doi.org/10.1159/000366130 |
||||
| 69 Patil CN, Wallace K, LaMarca BD, Moulana M, Lopez-Ruiz A, Soljancic A, Juncos LA, Grande JP, Reckelhoff JF: Low-dose testosterone protects against renal ischemia-reperfusion injury by increasing renal IL-10-to-TNF-α ratio and attenuating T-cell infiltration. Am J Physiol Renal Physiol 2016;311:F395-F403. https://doi.org/10.1152/ajprenal.00454.2015 |
||||
| 70 Deng J, Kohda Y, Chiao H, Wang Y, Hu X, Hewitt SM, Miyaji T, McLeroy P, Nibhanupudy B, Li S, Star RA: Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int 2001;60:2118-2128. https://doi.org/10.1046/j.1523-1755.2001.00043.x |
||||
| 71 Sakai K, Nozaki Y, Murao Y, Yano T, Ri J, Niki K, Kinoshita K, Funauchi M, Matsumura I: Protective effect, and mechanism of IL-10 on renal ischemia-reperfusion injury. Lab Invest 2019;99:671-683. https://doi.org/10.1038/s41374-018-0162-0 |
||||
| 72 Verkman AS: Aquaporins in endothelia. Kidney Int 2006;69:1120-1123. https://doi.org/10.1038/sj.ki.5000226 |
||||
| 73 Chebotareva N, Bobkova I, Shilov E: Heat shock proteins and kidney disease: perspectives of HSP therapy. Cell Stress Chaperones 2017;22:319-343. https://doi.org/10.1007/s12192-017-0790-0 |
||||
| 74 Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, Meng X: Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: the proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol Med 2014;20:280-289. https://doi.org/10.2119/molmed.2014.00058 |
||||
| 75 Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W, Cai W, Gao Y, Leak RK, Keep RF, Bennett MVL, Chen J: Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci USA 2017;114:E1243-E1252. https://doi.org/10.1073/pnas.1621174114 |
||||
| 76 Zhang HL, Jia KY, Sun D, Yang M: Protective effect of HSP27 in atherosclerosis and coronary heart disease by inhibiting reactive oxygen species. J Cell Biochem 2019;120:2859-2868. https://doi.org/10.1002/jcb.26575 |
||||
| 77 Xiong B, Li M, Xiang S, Han L: A1AR-mediated renal protection against ischemia/reperfusion injury is dependent on HSP27 induction. Int Urol Nephrol 2018;50:1355-1363. https://doi.org/10.1007/s11255-018-1797-x |
||||
| 78 He K, Chen X, Han C, Xu L, Zhang J, Zhang M, Xia Q: Lipopolysaccharide-induced cross-tolerance against renal ischemia-reperfusion injury is mediated by hypoxia-inducible factor-2α-regulated nitric oxide production. Kidney Int 2014;85:276-288. https://doi.org/10.1038/ki.2013.342 |
||||
| 79 He K, Xia L, Zhang J: LPS ameliorates renal ischemia/reperfusion injury via Hsp27 up-regulation. Int Urol Nephrol 2018;50:571-580. https://doi.org/10.1007/s11255-017-1735-3 |
||||
| 80 Zeeshan HM, Lee GH, Kim HR, Chae HJ: Endoplasmic Reticulum Stress and Associated ROS. Int J Mol Sci 2016;17:327. https://doi.org/10.3390/ijms17030327 |
||||