×
![]()
Corresponding Author: Nathalie Strutz-Seebohm and Guiscard Seebohm
Department of Cardiovascular Medicine, Institute for Genetics of Heart Diseases (IfGH), University Hospital Muenster, Robert‐Koch Straße 45, D‐48149 Muenster (Germany)
Tel. +49 (0)251/83‐58255, Fax +49 (0)251/83‐58257, E-Mail nathalie.strutz-seebohm@ukmuenster.de; guiscard.seebohm@ukmuenster.de
Virus-Host Interactions of Enteroviruses and Parvovirus B19 in Myocarditis
Huyen Tran Ho Stefan Peischard Nathalie Strutz-Seebohm Guiscard Seebohm
Cellular Electrophysiology and Molecular Biology, Institute for Genetics of Heart Diseases (IfGH), University Hospital Münster, Münster, Germany
Relevance of CVB3 and PVB19 in Viral Myocarditis
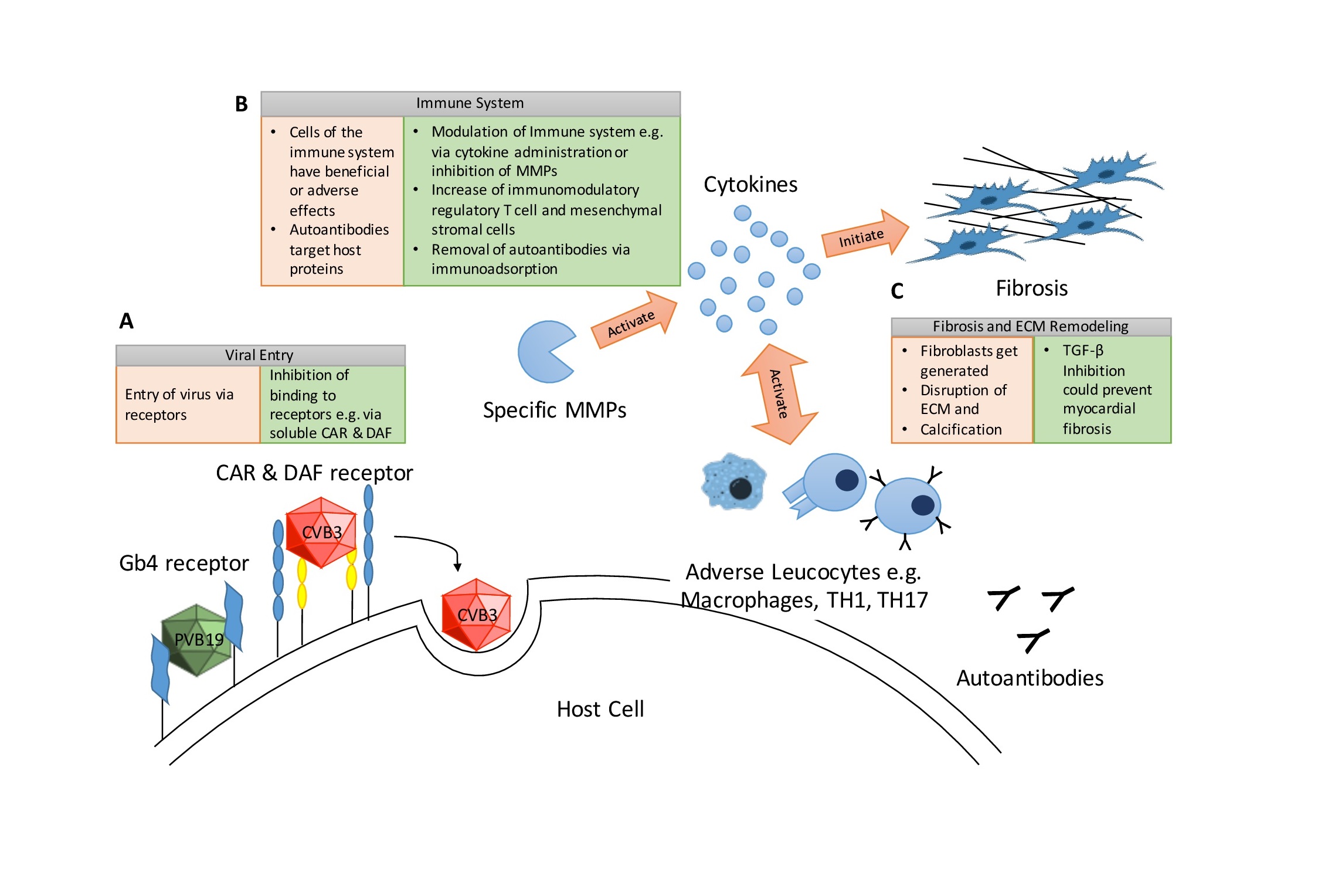
Myocarditis is the process of inflammation in the myocardium. One cause for myocarditis are viruses, that infect the host’s heart. The severity of viral myocarditis (VMC) varies between a light illness with flu-like symptoms and gastrointestinal illness to severe dilated cardiomyopathy (DCM) and sudden cardiac death [1]. In western countries the most prominent cause for myocarditis is chronic viral infection and evidence is emerging that also the newly emerged SARS-CoV-2 can cause myocarditis [2, 3]. Dadashi et al. recently conducted a thorough review and meta-analysis of the prevalence of most common viruses in viral myocarditis including 75 studies from 1973 to 2018 performed in highly industrialized nations. In conclusion, the viruses with the highest prevalence rates were parvovirus B19 (PVB19) with 25.0% and non-polio enteroviruses with 18% [4]. However, the relevance of PVB19 is questionable, as the prevalence of PVB19 in healthy hearts is similar to hearts with diagnosed myocarditis (MC) or DCM [5]. Anyhow, it is proposed that a high viral load of over 500 ge (genome equivalents) and active replication of PVB19 could result in myocarditis in about 64.7% of myocarditis patients [6]. Additional co-infection with another cardiotropic virus in parallel to a PVB19 infection might even elevate the chance of myocarditis development [7, 8]. Kuhl et al. were able to support this hypothesis: They observed a clear difference between latent and transcriptionally active PVB19 in human endomyocardial biopsies. Thus, the shift from latent PVB19 to transcriptionally active, potentially caused by transactivating viruses (e.g. human herpes virus 6) and/or immune suppression, can result in the emergence of myocardial dysfunction [9].
Since viruses are the main cause for myocarditis, it is important to investigate and find treatments against the viruses that most commonly cause myocarditis, namely PVB19 and enteroviruses. By understanding these viruses, parallels to other, even less understood viruses, might be drawn. Hence, this review focuses on the effects of enteroviruses such as Coxsackievirus B3 (CVB3) and PVB19 on the human heart in order to understand underlying mechanisms of the pathogenesis of viral myocarditis and to discuss novel treatment options. Since there is a current lack of efficient treatment and cure against myocarditis viruses [10, 11], it is even more important to understand the common modi operandi of the respective viruses.
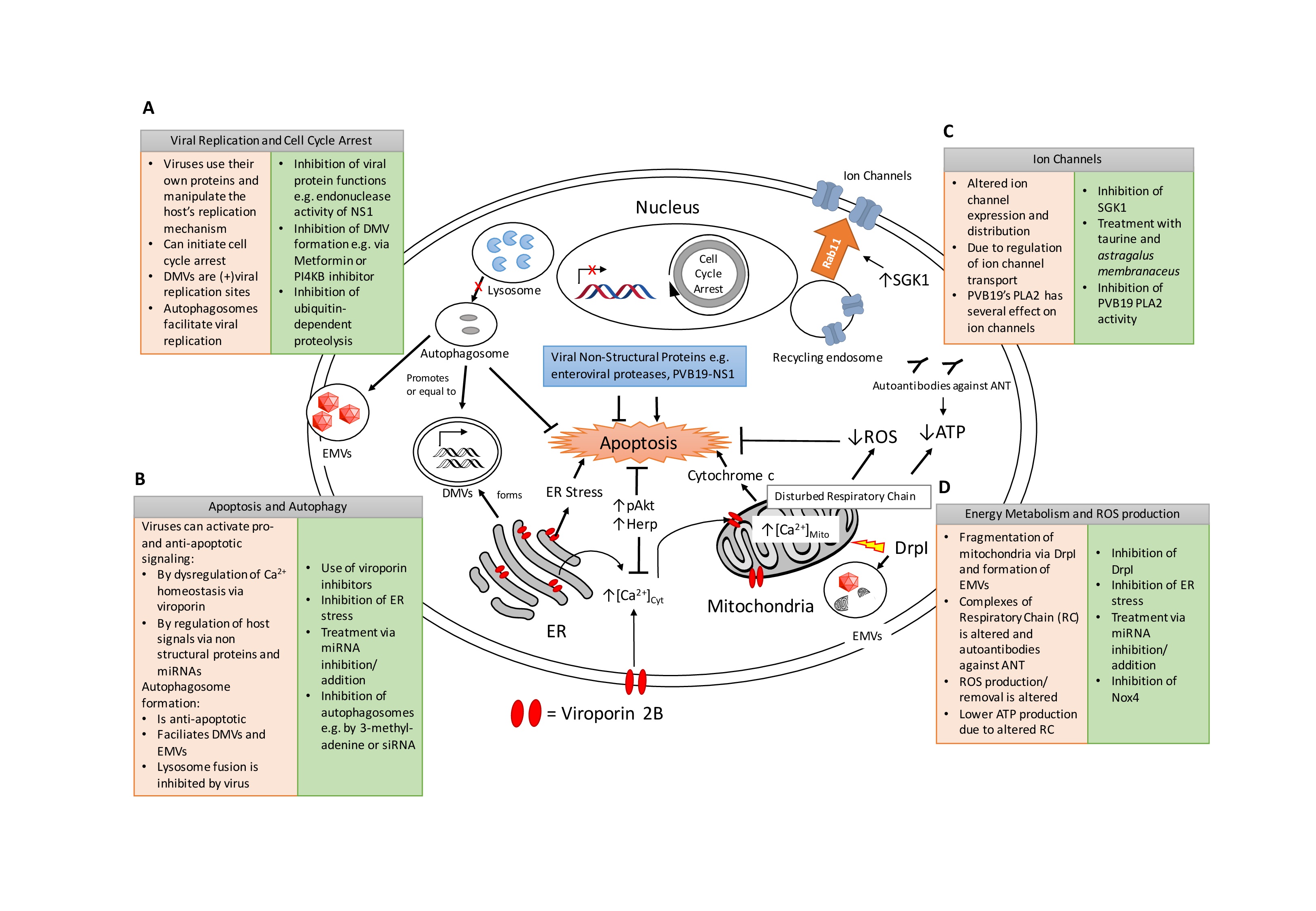
Viruses use the host cell to their advantage to reproduce and distribute viral particles by reprogramming intracellular mechanisms of the host cell. These mechanisms include immune responses, autophagy, formation of viral replication sites, cell cycle alterations, apoptosis, energy metabolism, and electrophysiological alterations that will be addressed in the next sections.
Viral genomes of Enteroviruses (Coxsackievirus B3) and Parvovirus B19
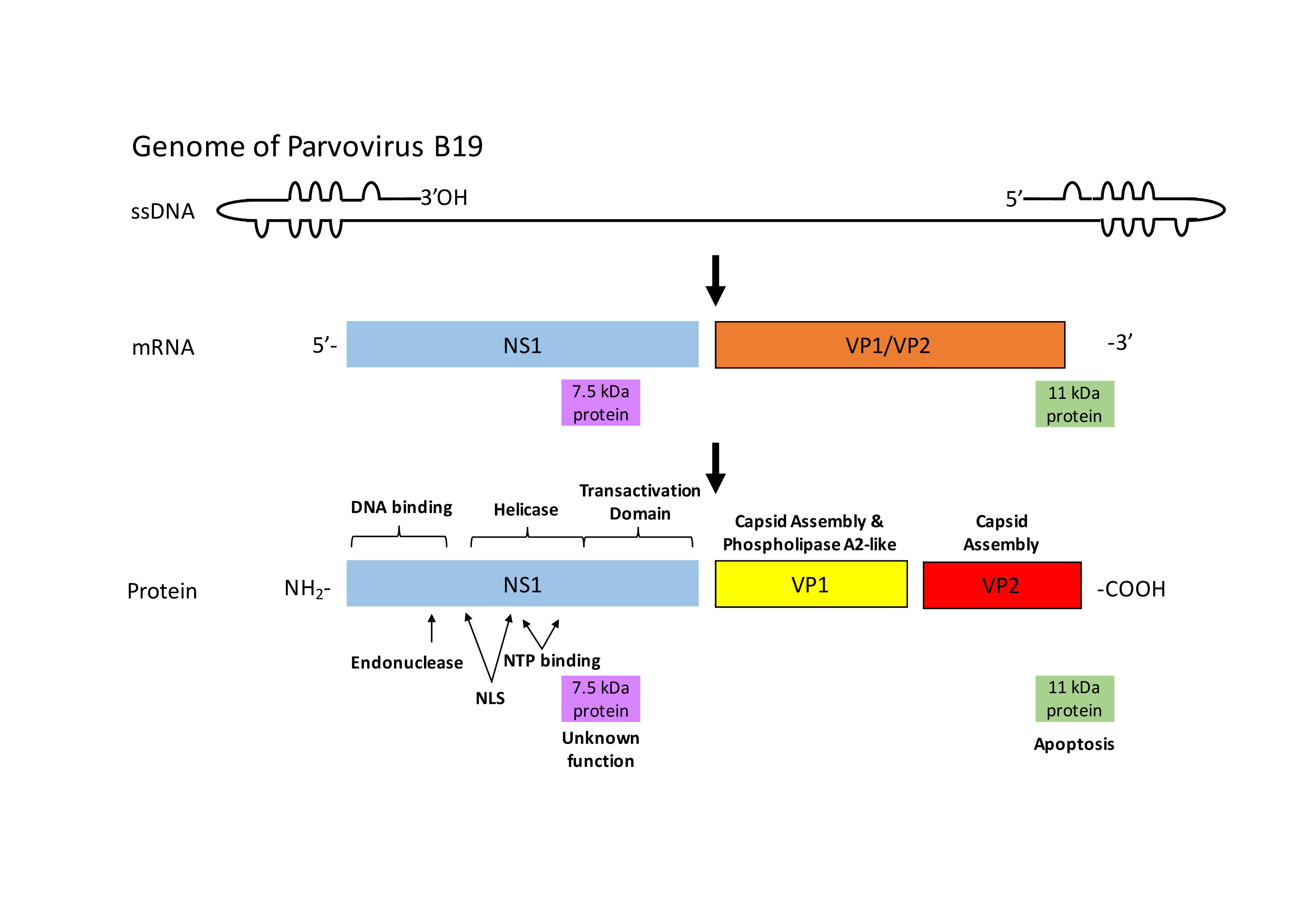
Viral genomes are customarily small and therefore code only for a few structural proteins forming the viral capsid and non-structural proteins which control the host cell-virus interactions. These interactions serve to promote viral replication and virus assembly and they vary depending on the virus type [12, 13]. PVB19 is an ssDNA virus while CVB3 is an ssRNA virus. Both are non-enveloped viruses and are considered comparatively small with genome sizes of 7.4 kb (CVB3) and 5.6 kb (PVB19) respectively [14, 15].
CVB3
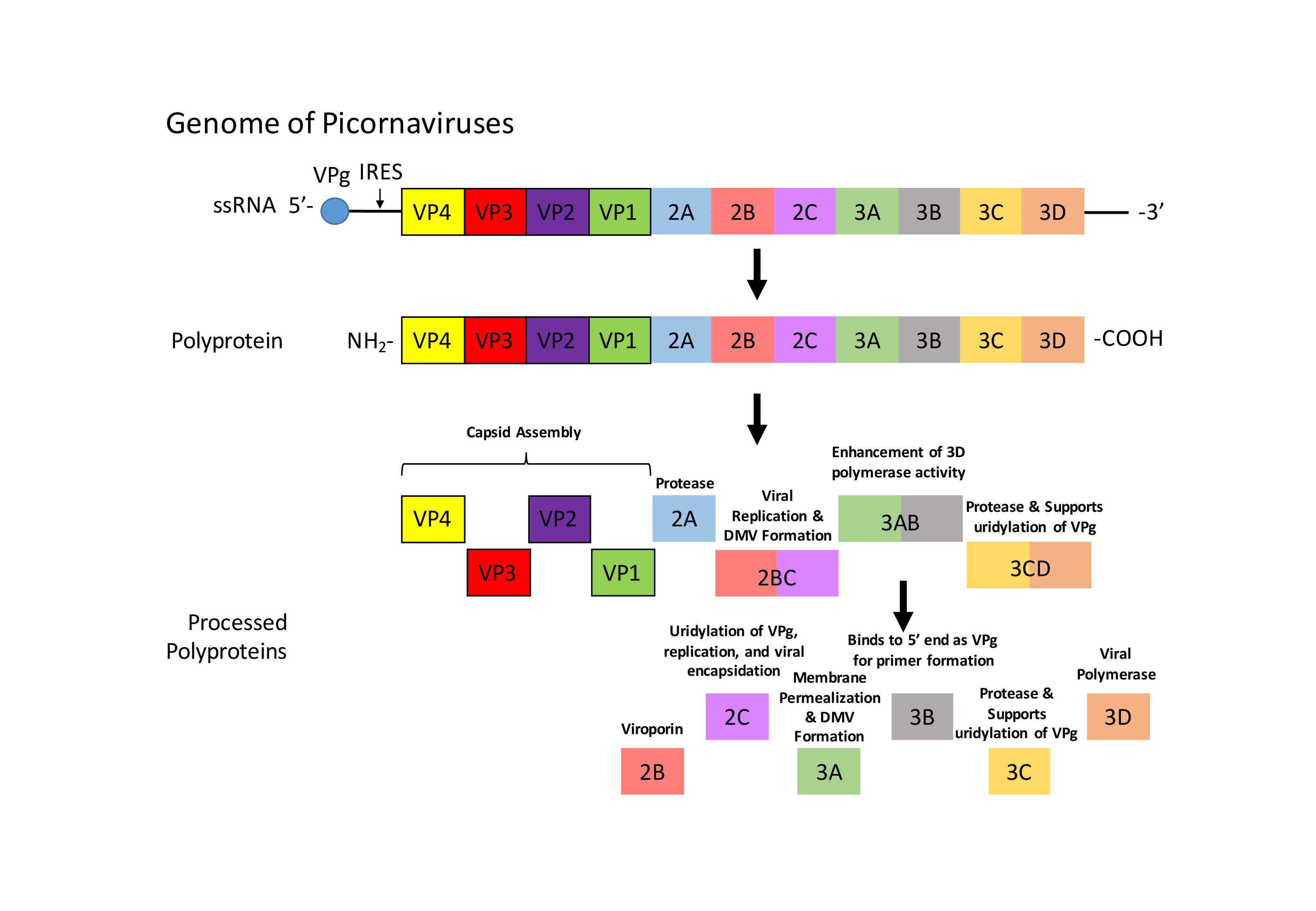
The picornaviral genome is coding for the structural proteins VP1-VP4 and for the non-structural proteins 2A-3D (Fig. 1) [16, 17]. As previously mentioned, the structural proteins form the capsid. Differential proteolysis of the polyprotein gives rise to non-structural proteins, such as viral proteases 2A, 3C, and 3CD, a viral polymerase (3D), a viroporin (2B), and proteins for viral encapsulation (2C), membrane permeabilization (3C), and viral replication (2BC) [17]. The genomic ssRNA is not capped at the 5’ end but the viral 3B protein, also called virion protein genome linked (VPg), is recruited to the 5’ end, which serves as a primer for viral replication [18]. Moreover, the CVB3 genome inherits an internal ribosome entry site (IRES) at the 5’ end that mediates translation of the viral RNA [19].
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG) under grant number DFG Se1077/13-1 to G.S.
The authors declare that no conflict of interests exist.
| 1 Esfandiarei M, McManus BM: Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol 2008;3:127-155. https://doi.org/10.1146/annurev.pathmechdis.3.121806.151534 |
||||
| 2 Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Heliö T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi L, et al.: Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013;34:2636-2648, 2648a-2648d. https://doi.org/10.1093/eurheartj/eht210 |
||||
| 3 Caforio AL, Calabrese F, Angelini A, Tona F, Vinci A, Bottaro S, Ramondo A, Carturan E, Iliceto S, Thiene G, Daliento L: A prospective study of biopsy-proven myocarditis: prognostic relevance of clinical and aetiopathogenetic features at diagnosis. Eur Heart J 2007;28:1326-1333. https://doi.org/10.1093/eurheartj/ehm076 |
||||
| 4 Dadashi M, Azimi T, Faghihloo E: Global study of viral myocarditis: A systematic review and meta-analysis. J Acute Dis 2020;9:1. | ||||
| 5 Verdonschot J, Hazebroek M, Merken J, Debing Y, Dennert R, Brunner-La Rocca HP, Heymans S: Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: review of the literature. Eur J Heart Fail 2016;18:1430-1441. https://doi.org/10.1002/ejhf.665 |
||||
| 6 Bock CT, Klingel K, Kandolf R: Human parvovirus B19-associated myocarditis. N Engl J Med 2010;362:1248-1249. https://doi.org/10.1056/NEJMc0911362 |
||||
| 7 Callon D, Berri F, Lebreil AL, Fornès P, Andreoletti L: Coinfection of Parvovirus B19 with Influenza A/H1N1 Causes Fulminant Myocarditis and Pneumonia. An Autopsy Case Report. Pathogens 2021;10:958. https://doi.org/10.3390/pathogens10080958 |
||||
| 8 Das BB, Prusty BK, Niu J, Huang ML, Zhu H, Eliassen E, Kuypers JM, Jerome KR: Detection of parvovirus B19 and human herpesvirus 6 in pediatric dilated cardiomyopathy: Impact after heart transplantation. Ann Pediatr Cardiol 2020;13:301-308. https://doi.org/10.4103/apc.APC_124_19 |
||||
| 9 Kuhl U, Lassner D, Dorner A, Rohde M, Escher F, Seeberg B, Hertel E, Tschope C, Skurk C, Gross UM, Schultheiss HP, Poller W: A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression. Basic Res Cardiol 2013;108:372. https://doi.org/10.1007/s00395-013-0372-y |
||||
| 10 Pollack A, Kontorovich AR, Fuster V, Dec GW: Viral myocarditis--diagnosis, treatment options, and current controversies. Nat Rev Cardiol 2015;12:670-680. https://doi.org/10.1038/nrcardio.2015.108 |
||||
| 11 Tschöpe C, Cooper LT, Torre-Amione G, Van Linthout S: Management of Myocarditis-Related Cardiomyopathy in Adults. Circ Res 2019;124:1568-1583. https://doi.org/10.1161/CIRCRESAHA.118.313578 |
||||
| 12 Gelderblom HR: Structure and Classification of Viruses (Chapter 41); in Baron S (eds): Medical Microbiology. 4th ed. Galveston (TX), University of Texas Medical Branch at Galveston, 1996. | ||||
| 13 Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R: Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog 2010;6:e1001233. https://doi.org/10.1371/journal.ppat.1001233 |
||||
| 14 Zhi N, Zádori Z, Brown KE, Tijssen P: Construction and sequencing of an infectious clone of the human parvovirus B19. Virology 2004;318:142-152. https://doi.org/10.1016/j.virol.2003.09.011 |
||||
| 15 Bowles NE, Richardson PJ, Olsen EG, Archard LC: Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1986;1:1120-1123. https://doi.org/10.1016/S0140-6736(86)91837-4 |
||||
| 16 Lindberg AM, Stålhandske PO, Pettersson U: Genome of coxsackievirus B3. Virology 1987;156:50-63. https://doi.org/10.1016/0042-6822(87)90435-1 |
||||
| 17 Buenz EJ, Howe CL: Picornaviruses and cell death. Trends Microbiol 2006;14:28-36. https://doi.org/10.1016/j.tim.2005.11.003 |
||||
| 18 Nayak A, Goodfellow IG, Belsham GJ: Factors required for the Uridylylation of the foot-and-mouth disease virus 3B1, 3B2, and 3B3 peptides by the RNA-dependent RNA polymerase (3Dpol) in vitro. J Virol 2005;79:7698-7706. https://doi.org/10.1128/JVI.79.12.7698-7706.2005 |
||||
| 19 Yang Y, Wang Z: IRES-mediated cap-independent translation, a path leading to hidden proteome. J Mol Cell Biol 2019;11:911-919. https://doi.org/10.1093/jmcb/mjz091 |
||||
| 20 Qiu J, Söderlund-Venermo M, Young NS: Human Parvoviruses. Clin Microbiol Rev 2017;30:43-113. https://doi.org/10.1128/CMR.00040-16 |
||||
| 21 Ozawa K, Young N: Characterization of capsid and noncapsid proteins of B19 parvovirus propagated in human erythroid bone marrow cell cultures. J Virol 1987;61:2627-2630. https://doi.org/10.1128/jvi.61.8.2627-2630.1987 |
||||
| 22 Ganaie SS, Qiu J: Recent Advances in Replication and Infection of Human Parvovirus B19. Front Cell Infect Microbiol 2018;8:166. https://doi.org/10.3389/fcimb.2018.00166 |
||||
| 23 Pillet S, Annan Z, Fichelson S, Morinet F: Identification of a nonconventional motif necessary for the nuclear import of the human parvovirus B19 major capsid protein (VP2). Virology 2003;306:25-32. https://doi.org/10.1016/S0042-6822(02)00047-8 |
||||
| 24 Chen AY, Zhang EY, Guan W, Cheng F, Kleiboeker S, Yankee TM, Qiu J: The small 11 kDa nonstructural protein of human parvovirus B19 plays a key role in inducing apoptosis during B19 virus infection of primary erythroid progenitor cells. Blood 2010;115:1070-1080. https://doi.org/10.1182/blood-2009-04-215756 |
||||
| 25 Cotmore SF, McKie VC, Anderson LJ, Astell CR, Tattersall P: Identification of the major structural and nonstructural proteins encoded by human parvovirus B19 and mapping of their genes by procaryotic expression of isolated genomic fragments. J Virol 1986;60:548-557. https://doi.org/10.1128/jvi.60.2.548-557.1986 |
||||
| 26 Bültmann BD, Klingel K, Sotlar K, Bock CT, Baba HA, Sauter M, Kandolf R: Fatal parvovirus B19-associated myocarditis clinically mimicking ischemic heart disease: an endothelial cell-mediated disease. Hum Pathol 2003;34:92-95. https://doi.org/10.1053/hupa.2003.48 |
||||
| 27 Favere K, Bosman M, Klingel K, Heymans S, Van Linthout S, Delputte PL, De Sutter J, Heidbuchel H, Guns PJ: Toll-Like Receptors: Are They Taking a Toll on the Heart in Viral Myocarditis? Viruses 2021;13:1003. https://doi.org/10.3390/v13061003 |
||||
| 28 Bergelson JM, Krithivas A, Celi L, Droguett G, Horwitz MS, Wickham T, Crowell RL, Finberg RW: The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J Virol 1998;72:415-419. https://doi.org/10.1128/JVI.72.1.415-419.1998 |
||||
| 29 Shafren DR, Williams DT, Barry RD: A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J Virol 1997;71:9844-9848. https://doi.org/10.1128/jvi.71.12.9844-9848.1997 |
||||
| 30 Pinkert S, Westermann D, Wang X, Klingel K, Dörner A, Savvatis K, Grössl T, Krohn S, Tschöpe C, Zeichhardt H, Kotsch K, Weitmann K, Hoffmann W, Schultheiss HP, Spiller OB, Poller W, Fechner H: Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation 2009;120:2358-2366. https://doi.org/10.1161/CIRCULATIONAHA.108.845339 |
||||
| 31 Yanagawa B, Spiller OB, Choy J, Luo H, Cheung P, Zhang HM, Goodfellow IG, Evans DJ, Suarez A, Yang D, McManus BM: Coxsackievirus B3-associated myocardial pathology and viral load reduced by recombinant soluble human decay-accelerating factor in mice. Lab Invest 2003;83:75-85. https://doi.org/10.1097/01.LAB.0000049349.56211.09 |
||||
| 32 Brown KE, Anderson SM, Young NS: Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science 1993;262:114-117. https://doi.org/10.1126/science.8211117 |
||||
| 33 Bieri J, Ros C: Globoside Is Dispensable for Parvovirus B19 Entry but Essential at a Postentry Step for Productive Infection. J Virol 2019;93:e00972-19. https://doi.org/10.1128/JVI.00972-19 |
||||
| 34 Okuda T, Nakakita S, Nakayama K: Structural characterization and dynamics of globotetraosylceramide in vascular endothelial cells under TNF-alpha stimulation. Glycoconj J 2010;27:287-296. https://doi.org/10.1007/s10719-009-9277-2 |
||||
| 35 Munakata Y, Saito-Ito T, Kumura-Ishii K, Huang J, Kodera T, Ishii T, Hirabayashi Y, Koyanagi Y, Sasaki T: Ku80 autoantigen as a cellular coreceptor for human parvovirus B19 infection. Blood 2005;106:3449-3456. https://doi.org/10.1182/blood-2005-02-0536 |
||||
| 36 Weigel-Kelley KA, Yoder MC, Srivastava A: Alpha5beta1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of beta1 integrin for viral entry. Blood 2003;102:3927-3933. https://doi.org/10.1182/blood-2003-05-1522 |
||||
| 37 von Kietzell K, Pozzuto T, Heilbronn R, Grössl T, Fechner H, Weger S: Antibody-mediated enhancement of parvovirus B19 uptake into endothelial cells mediated by a receptor for complement factor C1q. J Virol 2014;88:8102-8115. https://doi.org/10.1128/JVI.00649-14 |
||||
| 38 Munakata Y, Kato I, Saito T, Kodera T, Ishii KK, Sasaki T: Human parvovirus B19 infection of monocytic cell line U937 and antibody-dependent enhancement. Virology 2006;345:251-257. https://doi.org/10.1016/j.virol.2005.09.040 |
||||
| 39 Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU: Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med 1999;5:320-326. https://doi.org/10.1038/6543 |
||||
| 40 Kandolf R, Ameis D, Kirschner P, Canu A, Hofschneider PH: In situ detection of enteroviral genomes in myocardial cells by nucleic acid hybridization: an approach to the diagnosis of viral heart disease. Proc Natl Acad Sci U S A 1987;84:6272-6276. https://doi.org/10.1073/pnas.84.17.6272 |
||||
| 41 Mann DL: The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res 2011;108:1133-1145. https://doi.org/10.1161/CIRCRESAHA.110.226936 |
||||
| 42 Corsten MF, Schroen B, Heymans S: Inflammation in viral myocarditis: friend or foe? Trends Mol Med 2012;18:426-437. https://doi.org/10.1016/j.molmed.2012.05.005 |
||||
| 43 Xu M, Li X, Song L, Tao C, Fang J, Tao L: Lupeol alleviates coxsackievirus B3-induced viral myocarditis in mice via downregulating toll-like receptor 4. J Int Med Res 2020;48:300060520910908. https://doi.org/10.1177/0300060520910908 |
||||
| 44 Fei Y, Chaulagain A, Wang T, Chen Y, Liu J, Yi M, Wang Y, Huang Y, Lin L, Chen S, Xu W, Tong L, Wu X, Zhao D, Zhang F, Zhao W, Zhong Z: MiR-146a down-regulates inflammatory response by targeting TLR3 and TRAF6 in Coxsackievirus B infection. RNA 2020;26:91-100. https://doi.org/10.1261/rna.071985.119 |
||||
| 45 Nie S, Dong B, Gao S, Zhou Y, Lu W, Fang M, Hua S, Yu Y, Wang L: The protective effect of interfering TLR9-IRF5 signaling pathway on the development of CVB3-induced myocarditis. Clin Immunol 2019;207:24-35. https://doi.org/10.1016/j.clim.2019.07.002 |
||||
| 46 Kraft L, Erdenesukh T, Sauter M, Tschöpe C, Klingel K: Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol 2019;114:11. https://doi.org/10.1007/s00395-019-0719-0 |
||||
| 47 Wang YX, da Cunha V, Vincelette J, White K, Velichko S, Xu Y, Gross C, Fitch RM, Halks-Miller M, Larsen BR, Yajima T, Knowlton KU, Vergona R, Sullivan ME, Croze E: Antiviral and myocyte protective effects of murine interferon-beta and -{alpha}2 in coxsackievirus B3-induced myocarditis and epicarditis in Balb/c mice. Am J Physiol Heart Circ Physiol 2007;293:H69-76. https://doi.org/10.1152/ajpheart.00154.2007 |
||||
| 48 Heim A, Weiss S: Interferons in enteroviral heart disease: modulation of cytokine expression and antiviral activity. Med Microbiol Immunol 2004;193:149-154. https://doi.org/10.1007/s00430-003-0200-3 |
||||
| 49 Kühl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, Poller W, Schultheiss HP: Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 2003;107:2793-2798. https://doi.org/10.1161/01.CIR.0000072766.67150.51 |
||||
| 50 Kuhl U, Lassner D, von Schlippenbach J, Poller W, Schultheiss HP: Interferon-Beta improves survival in enterovirus-associated cardiomyopathy. J Am Coll Cardiol 2012;60:1295-1296. https://doi.org/10.1016/j.jacc.2012.06.026 |
||||
| 51 Horwitz MS, La Cava A, Fine C, Rodriguez E, Ilic A, Sarvetnick N: Pancreatic expression of interferon-gamma protects mice from lethal coxsackievirus B3 infection and subsequent myocarditis. Nat Med 2000;6:693-697. https://doi.org/10.1038/76277 |
||||
| 52 Wessely R, Klingel K, Knowlton KU, Kandolf R: Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation 2001;103:756-761. https://doi.org/10.1161/01.CIR.103.5.756 |
||||
| 53 Yasukawa H, Yajima T, Duplain H, Iwatate M, Kido M, Hoshijima M, Weitzman MD, Nakamura T, Woodard S, Xiong D, Yoshimura A, Chien KR, Knowlton KU: The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury. J Clin Invest 2003;111:469-478. https://doi.org/10.1172/JCI16491 |
||||
| 54 Yajima T, Yasukawa H, Jeon ES, Xiong D, Dorner A, Iwatate M, Nara M, Zhou H, Summers-Torres D, Hoshijima M, Chien KR, Yoshimura A, Knowlton KU: Innate defense mechanism against virus infection within the cardiac myocyte requiring gp130-STAT3 signaling. Circulation 2006;114:2364-2373. https://doi.org/10.1161/CIRCULATIONAHA.106.642454 |
||||
| 55 Lane JR, Neumann DA, Lafond-Walker A, Herskowitz A, Rose NR: Interleukin 1 or tumor necrosis factor can promote Coxsackie B3-induced myocarditis in resistant B10.A mice. J Exp Med 1992;175:1123-1129. https://doi.org/10.1084/jem.175.4.1123 |
||||
| 56 Huang CH, Vallejo JG, Kollias G, Mann DL: Role of the innate immune system in acute viral myocarditis. Basic Res Cardiol 2009;104:228-237. https://doi.org/10.1007/s00395-008-0765-5 |
||||
| 57 Wada H, Saito K, Kanda T, Kobayashi I, Fujii H, Fujigaki S, Maekawa N, Takatsu H, Fujiwara H, Sekikawa K, Seishima M: Tumor necrosis factor-alpha (TNF-alpha) plays a protective role in acute viralmyocarditis in mice: A study using mice lacking TNF-alpha. Circulation 2001;103:743-749. https://doi.org/10.1161/01.CIR.103.5.743 |
||||
| 58 Godeny EK, Gauntt CJ: Involvement of natural killer cells in coxsackievirus B3-induced murine myocarditis. J Immunol 1986;137:1695-1702. | ||||
| 59 Klingel K, Fabritius C, Sauter M, Göldner K, Stauch D, Kandolf R, Ettischer N, Gahlen S, Schönberger T, Ebner S, Makrigiannis AP, Bélanger S, Diefenbach A, Polić B, Pratschke J, Kotsch K: The activating receptor NKG2D of natural killer cells promotes resistance against enterovirus-mediated inflammatory cardiomyopathy. J Pathol 2014;234:164-177. https://doi.org/10.1002/path.4369 |
||||
| 60 Hirasawa K, Tsutsui S, Takeda M, Mizutani M, Itagaki S, Doi K: Depletion of Mac1-positive macrophages protects DBA/2 mice from encephalomyocarditis virus-induced myocarditis and diabetes. J Gen Virol 1996;77:737-741. https://doi.org/10.1099/0022-1317-77-4-737 |
||||
| 61 Yang X, Yue Y, Xiong S: Dpep2 Emerging as a Modulator of Macrophage Inflammation Confers Protection Against CVB3-Induced Viral Myocarditis. Front Cell Infect Microbiol 2019;9:57. https://doi.org/10.3389/fcimb.2019.00057 |
||||
| 62 Papageorgiou AP, Heymans S: Interactions between the extracellular matrix and inflammation during viral myocarditis. Immunobiology 2012;217:503-510. https://doi.org/10.1016/j.imbio.2011.07.011 |
||||
| 63 Page-McCaw A, Ewald AJ, Werb Z: Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 2007;8:221-233. https://doi.org/10.1038/nrm2125 |
||||
| 64 Westermann D, Savvatis K, Schultheiss HP, Tschöpe C: Immunomodulation and matrix metalloproteinases in viral myocarditis. J Mol Cell Cardiol 2010;48:468-473. https://doi.org/10.1016/j.yjmcc.2009.08.019 |
||||
| 65 Heymans S, Pauschinger M, De Palma A, Kallwellis-Opara A, Rutschow S, Swinnen M, Vanhoutte D, Gao F, Torpai R, Baker AH, Padalko E, Neyts J, Schultheiss HP, Van de Werf F, Carmeliet P, Pinto YM: Inhibition of urokinase-type plasminogen activator or matrix metalloproteinases prevents cardiac injury and dysfunction during viral myocarditis. Circulation 2006;114:565-573. https://doi.org/10.1161/CIRCULATIONAHA.105.591032 |
||||
| 66 Pauschinger M, Rutschow S, Chandrasekharan K, Westermann D, Weitz A, Peter Schwimmbeck L, Zeichhardt H, Poller W, Noutsias M, Li J, Schultheiss HP, Tschope C: Carvedilol improves left ventricular function in murine coxsackievirus-induced acute myocarditis association with reduced myocardial interleukin-1beta and MMP-8 expression and a modulated immune response. Eur J Heart Fail 2005;7:444-452. https://doi.org/10.1016/j.ejheart.2004.07.002 |
||||
| 67 Cheung C, Marchant D, Walker EK, Luo Z, Zhang J, Yanagawa B, Rahmani M, Cox J, Overall C, Senior RM, Luo H, McManus BM: Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice. Circulation 2008;117:1574-1582. https://doi.org/10.1161/CIRCULATIONAHA.107.733238 |
||||
| 68 Westermann D, Savvatis K, Lindner D, Zietsch C, Becher PM, Hammer E, Heimesaat MM, Bereswill S, Völker U, Escher F, Riad A, Plendl J, Klingel K, Poller W, Schultheiss HP, Tschöpe C: Reduced degradation of the chemokine MCP-3 by matrix metalloproteinase-2 exacerbates myocardial inflammation in experimental viral cardiomyopathy. Circulation 2011;124:2082-2093. https://doi.org/10.1161/CIRCULATIONAHA.111.035964 |
||||
| 69 Schmidt-Lucke C, Spillmann F, Bock T, Kühl U, Van Linthout S, Schultheiss HP, Tschöpe C: Interferon beta modulates endothelial damage in patients with cardiac persistence of human parvovirus b19 infection. J Infect Dis 2010;201:936-945. https://doi.org/10.1086/650700 |
||||
| 70 Schultheiss HP, Piper C, Sowade O, Waagstein F, Kapp JF, Wegscheider K, Groetzbach G, Pauschinger M, Escher F, Arbustini E, Siedentop H, Kuehl U: Betaferon in chronic viral cardiomyopathy (BICC) trial: Effects of interferon-β treatment in patients with chronic viral cardiomyopathy. Clin Res Cardiol 2016;105:763-773. https://doi.org/10.1007/s00392-016-0986-9 |
||||
| 71 Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R: Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci U S A 1992;89:314-318. https://doi.org/10.1073/pnas.89.1.314 |
||||
| 72 Klingel K, Rieger P, Mall G, Selinka HC, Huber M, Kandolf R: Visualization of enteroviral replication in myocardial tissue by ultrastructural in situ hybridization: identification of target cells and cytopathic effects. Lab Invest 1998;78:1227-1237. | ||||
| 73 Kaya Z, Leib C, Katus HA: Autoantibodies in heart failure and cardiac dysfunction. Circ Res 2012;110:145-158. https://doi.org/10.1161/CIRCRESAHA.111.243360 |
||||
| 74 Felix SB, Staudt A: Non-specific immunoadsorption in patients with dilated cardiomyopathy: mechanisms and clinical effects. Int J Cardiol 2006;112:30-33. https://doi.org/10.1016/j.ijcard.2006.05.014 |
||||
| 75 Staudt A, Hummel A, Ruppert J, Dörr M, Trimpert C, Birkenmeier K, Krieg T, Staudt Y, Felix SB: Immunoadsorption in dilated cardiomyopathy: 6-month results from a randomized study. Am Heart J 2006;152:712.e711-716. https://doi.org/10.1016/j.ahj.2006.06.027 |
||||
| 76 Doesch AO, Mueller S, Nelles M, Konstandin M, Celik S, Frankenstein L, Goeser S, Kaya Z, Koch A, Zugck C, Katus HA: Impact of troponin I-autoantibodies in chronic dilated and ischemic cardiomyopathy. Basic Res Cardiol 2011;106:25-35. https://doi.org/10.1007/s00395-010-0126-z |
||||
| 77 Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong JH, Wang JP, Zhang JH, Wang M, Guo HP, Cheng X, Liao YH: Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis. J Immunol 2010;185:4004-4010. https://doi.org/10.4049/jimmunol.1001718 |
||||
| 78 Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong JH, Wang JP, Zhang JH, Wang M, Guo HP, Liao YH: Neutralization of IL-17 inhibits the production of anti-ANT autoantibodies in CVB3-induced acute viral myocarditis. Int Immunopharmacol 2010;10:272-276. https://doi.org/10.1016/j.intimp.2009.11.010 |
||||
| 79 Myers JM, Cooper LT, Kem DC, Stavrakis S, Kosanke SD, Shevach EM, Fairweather D, Stoner JA, Cox CJ, Cunningham MW: Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight 2016;1:e85851. https://doi.org/10.1172/jci.insight.85851 |
||||
| 80 Wei H, Lin CK, Lu SJ, Wen YX, Yuan S, Liu YL: CD11b is involved in coxsackievirus B3-induced viral myocarditis in mice by inducing Th17 cells. Open Life Sci 2020;15:1024-1032. https://doi.org/10.1515/biol-2020-0085 |
||||
| 81 Pappritz K, Savvatis K, Miteva K, Kerim B, Dong F, Fechner H, Müller I, Brandt C, Lopez B, González A, Ravassa S, Klingel K, Diez J, Reinke P, Volk HD, Van Linthout S, Tschöpe C: Immunomodulation by adoptive regulatory T-cell transfer improves Coxsackievirus B3-induced myocarditis. FASEB J 2018; DOI: 10.1096/fj.201701408R. https://doi.org/10.1096/fj.201701408R |
||||
| 82 Koch M, Savvatis K, Scheeler M, Dhayat S, Bonaventura K, Pohl T, Riad A, Bulfone-Paus S, Schultheiss HP, Tschöpe C: Immunosuppression with an interleukin-2 fusion protein leads to improved LV function in experimental ischemic cardiomyopathy. Int Immunopharmacol 2010;10:207-212. https://doi.org/10.1016/j.intimp.2009.11.001 |
||||
| 83 Miteva K, Pappritz K, El-Shafeey M, Dong F, Ringe J, Tschöpe C, Van Linthout S: Mesenchymal Stromal Cells Modulate Monocytes Trafficking in Coxsackievirus B3-Induced Myocarditis. Stem Cells Transl Med 2017;6:1249-1261. https://doi.org/10.1002/sctm.16-0353 |
||||
| 84 Dominguez F, Kuhl U, Pieske B, Garcia-Pavia P, Tschope C: Update on Myocarditis and Inflammatory Cardiomyopathy: Reemergence of Endomyocardial Biopsy. Rev Esp Cardiol (Engl Ed) 2016;69:178-187. https://doi.org/10.1016/j.rec.2015.10.015 |
||||
| 85 Bujak M, Frangogiannis NG: The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 2007;74:184-195. https://doi.org/10.1016/j.cardiores.2006.10.002 |
||||
| 86 Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, Hunziker L, Matter CM, Eriksson U: Heart-infiltrating prominin-1+/CD133+ progenitor cells represent the cellular source of transforming growth factor beta-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circ Res 2009;105:462-470. https://doi.org/10.1161/CIRCRESAHA.109.196287 |
||||
| 87 Kania G, Blyszczuk P, Eriksson U: Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med 2009;19:247-252. https://doi.org/10.1016/j.tcm.2010.02.005 |
||||
| 88 Stallion A, Rafferty JF, Warner BW, Ziegler MM, Ryckman FC: Myocardial calcification: a predictor of poor outcome for myocarditis treated with extracorporeal life support. J Pediatr Surg 1994;29:492-494. https://doi.org/10.1016/0022-3468(94)90074-4 |
||||
| 89 Cooper LT: Myocarditis. N Engl J Med 2009;360:1526-1538. https://doi.org/10.1056/NEJMra0800028 |
||||
| 90 Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, Hare JM, Heidecker B, Heymans S, Hübner N, Kelle S, Klingel K, Maatz H, Parwani AS, Spillmann F, Starling RC, Tsutsui H, Seferovic P, Van Linthout S: Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol 2021;18:169-193. https://doi.org/10.1038/s41569-020-00435-x |
||||
| 91 Sinagra G, Porcari A, Gentile P, Artico J, Fabris E, Bussani R, Merlo M: Viral presence-guided immunomodulation in lymphocytic myocarditis: an update. Eur J Heart Fail 2021;23:211-216. https://doi.org/10.1002/ejhf.1969 |
||||
| 92 Tschöpe C, Van Linthout S, Jäger S, Arndt R, Trippel T, Müller I, Elsanhoury A, Rutschow S, Anker SD, Schultheiss HP, Pauschinger M, Spillmann F, Pappritz K: Modulation of the acute defence reaction by eplerenone prevents cardiac disease progression in viral myocarditis. ESC Heart Fail 2020;7:2838-2852. https://doi.org/10.1002/ehf2.12887 |
||||
| 93 Lee WS, Erdelyi K, Matyas C, Mukhopadhyay P, Varga ZV, Liaudet L, Haskú G, Čiháková D, Mechoulam R, Pacher P: Cannabidiol Limits T Cell-Mediated Chronic Autoimmune Myocarditis: Implications to Autoimmune Disorders and Organ Transplantation. Mol Med 2016;22:136-146. https://doi.org/10.2119/molmed.2016.00007 |
||||
| 94 Kazatchkine MD, Kaveri SV: Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med 2001;345:747-755. https://doi.org/10.1056/NEJMra993360 |
||||
| 95 Hazebroek MR, Henkens MTHM, Raafs AG, Verdonschot JAJ, Merken JJ, Dennert RM, Eurlings C, Abdul Hamid MA, Wolffs PFG, Winkens B, Brunner-La Rocca HP, Knackstedt C, van Paassen P, van Empel VPM, Heymans SRB: Intravenous immunoglobulin therapy in adult patients with idiopathic chronic cardiomyopathy and cardiac parvovirus B19 persistence: a prospective, double-blind, randomized, placebo-controlled clinical trial. Eur J Heart Fail 2021;23:302-309. https://doi.org/10.1002/ejhf.2082 |
||||
| 96 Mohamud Y, Shi J, Qu J, Poon T, Xue YC, Deng H, Zhang J, Luo H: Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Rep 2018;22:3292-3303. https://doi.org/10.1016/j.celrep.2018.02.090 |
||||
| 97 Huang L, Yue J: The interplay of autophagy and enterovirus. Semin Cell Dev Biol 2020;101:12-19. https://doi.org/10.1016/j.semcdb.2019.08.001 |
||||
| 98 Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, Nguyen DP, Sayen MR, Hilton BJ, Doran KS, Segall AM, Wolkowicz R, Cornell CT, Whitton JL, Gottlieb RA, Feuer R: Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog 2014;10:e1004045. https://doi.org/10.1371/journal.ppat.1004045 |
||||
| 99 Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H: Autophagosome supports coxsackievirus B3 replication in host cells. J Virol 2008;82:9143-9153. https://doi.org/10.1128/JVI.00641-08 |
||||
| 100 Tabor-Godwin JM, Tsueng G, Sayen MR, Gottlieb RA, Feuer R: The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 2012;8:938-953. https://doi.org/10.4161/auto.19781 |
||||
| 101 van der Schaar HM, Melia CE, van Bruggen JA, Strating JR, van Geenen ME, Koster AJ, Bárcena M, van Kuppeveld FJ: Illuminating the Sites of Enterovirus Replication in Living Cells by Using a Split-GFP-Tagged Viral Protein. mSphere 2016;1:e00104-16. https://doi.org/10.1128/mSphere.00104-16 |
||||
| 102 Jackson WT, Giddings TH, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K: Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005;3:e156. https://doi.org/10.1371/journal.pbio.0030156 |
||||
| 103 Harak C, Lohmann V: Ultrastructure of the replication sites of positive-strand RNA viruses. Virology 2015;479-480:418-433. https://doi.org/10.1016/j.virol.2015.02.029 |
||||
| 104 Wolff G, Melia CE, Snijder EJ, Bárcena M: Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol 2020;28:1022-1033. https://doi.org/10.1016/j.tim.2020.05.009 |
||||
| 105 Kirkegaard K, Jackson WT: Topology of double-membraned vesicles and the opportunity for non-lytic release of cytoplasm. Autophagy 2005;1:182-184. https://doi.org/10.4161/auto.1.3.2065 |
||||
| 106 Li X, Wang M, Cheng A, Wen X, Ou X, Mao S, Gao Q, Sun D, Jia R, Yang Q, Wu Y, Zhu D, Zhao X, Chen S, Liu M, Zhang S, Liu Y, Yu Y, Zhang L, Tian B, et al.: Enterovirus Replication Organelles and Inhibitors of Their Formation. Front Microbiol 2020;11:1817. https://doi.org/10.3389/fmicb.2020.01817 |
||||
| 107 Limpens RW, van der Schaar HM, Kumar D, Koster AJ, Snijder EJ, van Kuppeveld FJ, Bárcena M: The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. mBio 2011;2:e00166-11. https://doi.org/10.1128/mBio.00166-11 |
||||
| 108 Melia CE, Peddie CJ, de Jong AWM, Snijder EJ, Collinson LM, Koster AJ, van der Schaar HM, van Kuppeveld FJM, Bárcena M: Origins of Enterovirus Replication Organelles Established by Whole-Cell Electron Microscopy. mBio 2019;10:e00951-19. https://doi.org/10.1128/mBio.00951-19 |
||||
| 109 Suhy DA, Giddings TH, Kirkegaard K: Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol 2000;74:8953-8965. https://doi.org/10.1128/JVI.74.19.8953-8965.2000 |
||||
| 110 Alirezaei M, Flynn CT, Wood MR, Harkins S, Whitton JL: Coxsackievirus can exploit LC3 in both autophagy-dependent and -independent manners in vivo. Autophagy 2015;11:1389-1407. https://doi.org/10.1080/15548627.2015.1063769 |
||||
| 111 Melia CE, van der Schaar HM, Lyoo H, Limpens RWAL, Feng Q, Wahedi M, Overheul GJ, van Rij RP, Snijder EJ, Koster AJ, Bárcena M, van Kuppeveld FJM: Escaping Host Factor PI4KB Inhibition: Enterovirus Genomic RNA Replication in the Absence of Replication Organelles. Cell Rep 2017;21:587-599. https://doi.org/10.1016/j.celrep.2017.09.068 |
||||
| 112 Xie W, Wang L, Dai Q, Yu H, He X, Xiong J, Sheng H, Zhang D, Xin R, Qi Y, Hu F, Guo S, Zhang K: Activation of AMPK restricts coxsackievirus B3 replication by inhibiting lipid accumulation. J Mol Cell Cardiol 2015;85:155-167. https://doi.org/10.1016/j.yjmcc.2015.05.021 |
||||
| 113 Feuer R, Pagarigan RR, Harkins S, Liu F, Hunziker IP, Whitton JL: Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J Neurosci 2005;25:2434-2444. https://doi.org/10.1523/JNEUROSCI.4517-04.2005 |
||||
| 114 Luo H, Zhang J, Dastvan F, Yanagawa B, Reidy MA, Zhang HM, Yang D, Wilson JE, McManus BM: Ubiquitin-dependent proteolysis of cyclin D1 is associated with coxsackievirus-induced cell growth arrest. J Virol 2003;77:1-9. https://doi.org/10.1128/JVI.77.1.1-9.2003 |
||||
| 115 Luo H, Zhang J, Cheung C, Suarez A, McManus BM, Yang D: Proteasome inhibition reduces coxsackievirus B3 replication in murine cardiomyocytes. Am J Pathol 2003;163:381-385. https://doi.org/10.1016/S0002-9440(10)63667-X |
||||
| 116 Si X, McManus BM, Zhang J, Yuan J, Cheung C, Esfandiarei M, Suarez A, Morgan A, Luo H: Pyrrolidine dithiocarbamate reduces coxsackievirus B3 replication through inhibition of the ubiquitin-proteasome pathway. J Virol 2005;79:8014-8023. https://doi.org/10.1128/JVI.79.13.8014-8023.2005 |
||||
| 117 Wang Y, Zhao S, Chen Y, Wang T, Dong C, Wo X, Zhang J, Dong Y, Xu W, Feng X, Qu C, Wang Y, Zhong Z, Zhao W: The Capsid Protein VP1 of Coxsackievirus B Induces Cell Cycle Arrest by Up-Regulating Heat Shock Protein 70. Front Microbiol 2019;10:1633. https://doi.org/10.3389/fmicb.2019.01633 |
||||
| 118 Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL: Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 2002;76:4430-4440. https://doi.org/10.1128/JVI.76.9.4430-4440.2002 |
||||
| 119 Tewary SK, Zhao H, Deng X, Qiu J, Tang L: The human parvovirus B19 non-structural protein 1 N-terminal domain specifically binds to the origin of replication in the viral DNA. Virology 2014;449:297-303. https://doi.org/10.1016/j.virol.2013.11.031 |
||||
| 120 Xu P, Ganaie SS, Wang X, Wang Z, Kleiboeker S, Horton NC, Heier RF, Meyers MJ, Tavis JE, Qiu J: Endonuclease Activity Inhibition of the NS1 Protein of Parvovirus B19 as a Novel Target for Antiviral Drug Development. Antimicrob Agents Chemother 2019;63:e01879-18. https://doi.org/10.1128/AAC.01879-18 |
||||
| 121 Luo Y, Kleiboeker S, Deng X, Qiu J: Human parvovirus B19 infection causes cell cycle arrest of human erythroid progenitors at late S phase that favors viral DNA replication. J Virol 2013;87:12766-12775. https://doi.org/10.1128/JVI.02333-13 |
||||
| 122 Baggen J, Thibaut HJ, Strating JRPM, van Kuppeveld FJM: The life cycle of non-polio enteroviruses and how to target it. Nat Rev Microbiol 2018;16:368-381. https://doi.org/10.1038/s41579-018-0005-4 |
||||
| 123 Lai Y, Wang M, Cheng A, Mao S, Ou X, Yang Q, Wu Y, Jia R, Liu M, Zhu D, Chen S, Zhang S, Zhao XX, Huang J, Gao Q, Wang Y, Xu Z, Chen Z, Zhu L, Luo Q, et al.: Regulation of Apoptosis by Enteroviruses. Front Microbiol 2020;11:1145. https://doi.org/10.3389/fmicb.2020.01145 |
||||
| 124 Croft SN, Walker EJ, Ghildyal R: Picornaviruses and Apoptosis: Subversion of Cell Death. mBio 2017;8:e01009-17. https://doi.org/10.1128/mBio.01009-17 |
||||
| 125 Moffatt S, Yaegashi N, Tada K, Tanaka N, Sugamura K: Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J Virol 1998;72:3018-3028. https://doi.org/10.1128/JVI.72.4.3018-3028.1998 |
||||
| 126 Poole BD, Zhou J, Grote A, Schiffenbauer A, Naides SJ: Apoptosis of liver-derived cells induced by parvovirus B19 nonstructural protein. J Virol 2006;80:4114-4121. https://doi.org/10.1128/JVI.80.8.4114-4121.2006 |
||||
| 127 Hsu TC, Wu WJ, Chen MC, Tsay GJ: Human parvovirus B19 non-structural protein (NS1) induces apoptosis through mitochondria cell death pathway in COS-7 cells. Scand J Infect Dis 2004;36:570-577. https://doi.org/10.1080/00365540410016230 |
||||
| 128 Feng Q, Langereis MA, Lork M, Nguyen M, Hato SV, Lanke K, Emdad L, Bhoopathi P, Fisher PB, Lloyd RE, van Kuppeveld FJ: Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J Virol 2014;88:3369-3378. https://doi.org/10.1128/JVI.02712-13 |
||||
| 129 Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB: The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 2011;7:e1001311. https://doi.org/10.1371/journal.ppat.1001311 |
||||
| 130 Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z: Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 2013;9:e1003231. https://doi.org/10.1371/journal.ppat.1003231 |
||||
| 131 Esfandiarei M, Luo H, Yanagawa B, Suarez A, Dabiri D, Zhang J, McManus BM: Protein kinase B/Akt regulates coxsackievirus B3 replication through a mechanism which is not caspase dependent. J Virol 2004;78:4289-4298. https://doi.org/10.1128/JVI.78.8.4289-4298.2004 |
||||
| 132 Autret A, Martin-Latil S, Brisac C, Mousson L, Colbère-Garapin F, Blondel B: Early phosphatidylinositol 3-kinase/Akt pathway activation limits poliovirus-induced JNK-mediated cell death. J Virol 2008;82:3796-3802. https://doi.org/10.1128/JVI.02020-07 |
||||
| 133 Zhang H, Li F, Pan Z, Wu Z, Wang Y, Cui Y: Activation of PI3K/Akt pathway limits JNK-mediated apoptosis during EV71 infection. Virus Res 2014;192:74-84. https://doi.org/10.1016/j.virusres.2014.07.026 |
||||
| 134 Mirabelli C, Pelletier I, Téoulé F, Vidalain PO, Brisac C, Tangy F, Delpeyroux F, Blondel B: The CREB3-Herp signalling module limits the cytosolic calcium concentration increase and apoptosis induced by poliovirus. J Gen Virol 2016;97:2194-2200. https://doi.org/10.1099/jgv.0.000544 |
||||
| 135 Neznanov N, Kondratova A, Chumakov KM, Angres B, Zhumabayeva B, Agol VI, Gudkov AV: Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J Virol 2001;75:10409-10420. https://doi.org/10.1128/JVI.75.21.10409-10420.2001 |
||||
| 136 Xin L, Xiao Z, Ma X, He F, Yao H, Liu Z: Coxsackievirus B3 induces crosstalk between autophagy and apoptosis to benefit its release after replicating in autophagosomes through a mechanism involving caspase cleavage of autophagy-related proteins. Infect Genet Evol 2014;26:95-102. https://doi.org/10.1016/j.meegid.2014.05.005 |
||||
| 137 Li M, Wang X, Yu Y, Xie Y, Zou Y, Ge J, Peng T, Chen R: Coxsackievirus B3-induced calpain activation facilitates the progeny virus replication via a likely mechanism related with both autophagy enhancement and apoptosis inhibition in the early phase of infection: an in vitro study in H9c2 cells. Virus Res 2014;179:177-186. https://doi.org/10.1016/j.virusres.2013.10.014 |
||||
| 138 Song F, Yu X, Zhong T, Wang Z, Meng X, Li Z, Zhang S, Huo W, Liu X, Zhang Y, Zhang W, Yu J: Caspase-3 Inhibition Attenuates the Cytopathic Effects of EV71 Infection. Front Microbiol 2018;9:817. https://doi.org/10.3389/fmicb.2018.00817 |
||||
| 139 Li ML, Hsu TA, Chen TC, Chang SC, Lee JC, Chen CC, Stollar V, Shih SR: The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 2002;293:386-395. https://doi.org/10.1006/viro.2001.1310 |
||||
| 140 Li J, Yao Y, Chen Y, Xu X, Lin Y, Yang Z, Qiao W, Tan J: Enterovirus 71 3C Promotes Apoptosis through Cleavage of PinX1, a Telomere Binding Protein. J Virol 2017;91:e02016-16 https://doi.org/10.1128/JVI.02016-16 |
||||
| 141 Li ML, Lin JY, Chen BS, Weng KF, Shih SR, Calderon JD, Tolbert BS, Brewer G: EV71 3C protease induces apoptosis by cleavage of hnRNP A1 to promote apaf-1 translation. PLoS One 2019;14:e0221048. https://doi.org/10.1371/journal.pone.0221048 |
||||
| 142 Chau DH, Yuan J, Zhang H, Cheung P, Lim T, Liu Z, Sall A, Yang D: Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007;12:513-524. https://doi.org/10.1007/s10495-006-0013-0 |
||||
| 143 Hanson PJ, Ye X, Qiu Y, Zhang HM, Hemida MG, Wang F, Lim T, Gu A, Cho B, Kim H, Fung G, Granville DJ, Yang D: Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ 2016;23:828-840. https://doi.org/10.1038/cdd.2015.145 |
||||
| 144 Melton JV, Ewart GD, Weir RC, Board PG, Lee E, Gage PW: Alphavirus 6K proteins form ion channels. J Biol Chem 2002;277:46923-46931. https://doi.org/10.1074/jbc.M207847200 |
||||
| 145 Ewart GD, Sutherland T, Gage PW, Cox GB: The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol 1996;70:7108-7115. https://doi.org/10.1128/jvi.70.10.7108-7115.1996 |
||||
| 146 van Kuppeveld FJ, Hoenderop JG, Smeets RL, Willems PH, Dijkman HB, Galama JM, Melchers WJ: Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J 1997;16:3519-3532. https://doi.org/10.1093/emboj/16.12.3519 |
||||
| 147 Campanella M, de Jong AS, Lanke KW, Melchers WJ, Willems PH, Pinton P, Rizzuto R, van Kuppeveld FJ: The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J Biol Chem 2004;279:18440-18450. https://doi.org/10.1074/jbc.M309494200 |
||||
| 148 Putney JW, Jr.: A model for receptor-regulated calcium entry. Cell Calcium 1986;7:1-12. https://doi.org/10.1016/0143-4160(86)90026-6 |
||||
| 149 Putney JW, Jr.: Store-operated calcium channels: how do we measure them, and why do we care? Sci STKE 2004;2004:pe37. https://doi.org/10.1126/stke.2432004pe37 |
||||
| 150 Zhou Y, Frey TK, Yang JJ: Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 2009;46:1-17. https://doi.org/10.1016/j.ceca.2009.05.005 |
||||
| 151 Williams GS, Boyman L, Chikando AC, Khairallah RJ, Lederer WJ: Mitochondrial calcium uptake. Proc Natl Acad Sci U S A 2013;110:10479-10486. https://doi.org/10.1073/pnas.1300410110 |
||||
| 152 Tarasov AI, Griffiths EJ, Rutter GA: Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 2012;52:28-35. https://doi.org/10.1016/j.ceca.2012.03.003 |
||||
| 153 Li Z, Zou Z, Jiang Z, Huang X, Liu Q: Biological Function and Application of Picornaviral 2B Protein: A New Target for Antiviral Drug Development. Viruses 2019;11:510. https://doi.org/10.3390/v11060510 |
||||
| 154 Ow YP, Green DR, Hao Z, Mak TW: Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol 2008;9:532-542. https://doi.org/10.1038/nrm2434 |
||||
| 155 Cai Z, Shen L, Ma H, Yang J, Yang D, Chen H, Wei J, Lu Q, Wang DW, Xiang M, Wang J: Involvement of Endoplasmic Reticulum Stress-Mediated C/EBP Homologous Protein Activation in Coxsackievirus B3-Induced Acute Viral Myocarditis. Circ Heart Fail 2015;8:809-818. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001244 |
||||
| 156 Fung TS, Torres J, Liu DX: The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection. Viruses 2015;7:2834-2857. https://doi.org/10.3390/v7062749 |
||||
| 157 Han X, Cong H: Enterovirus 71 induces apoptosis by directly modulating the conformational activation of pro-apoptotic protein Bax. J Gen Virol 2017;98:422-434. https://doi.org/10.1099/jgv.0.000705 |
||||
| 158 Wang H, Li Y: Recent Progress on Functional Genomics Research of Enterovirus 71. Virol Sin 2019;34:9-21. https://doi.org/10.1007/s12250-018-0071-9 |
||||
| 159 Jiang D, Li M, Yu Y, Shi H, Chen R: microRNA-34a aggravates coxsackievirus B3-induced apoptosis of cardiomyocytes through the SIRT1-p53 pathway. J Med Virol 2019;91:1643-1651. https://doi.org/10.1002/jmv.25482 |
||||
| 160 Du X, Wang H, Xu F, Huang Y, Liu Z, Liu T: Enterovirus 71 induces apoptosis of SH‑SY5Y human neuroblastoma cells through stimulation of endogenous microRNA let-7b expression. Mol Med Rep 2015;12:953-959. https://doi.org/10.3892/mmr.2015.3482 |
||||
| 161 Chang YL, Ho BC, Sher S, Yu SL, Yang PC: miR-146a and miR-370 coordinate enterovirus 71-induced cell apoptosis through targeting SOS1 and GADD45β. Cell Microbiol 2015;17:802-818. https://doi.org/10.1111/cmi.12401 |
||||
| 162 Carthy CM, Granville DJ, Watson KA, Anderson DR, Wilson JE, Yang D, Hunt DW, McManus BM: Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. J Virol 1998;72:7669-7675. https://doi.org/10.1128/JVI.72.9.7669-7675.1998 |
||||
| 163 Carthy CM, Yanagawa B, Luo H, Granville DJ, Yang D, Cheung P, Cheung C, Esfandiarei M, Rudin CM, Thompson CB, Hunt DW, McManus BM: Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology 2003;313:147-157. https://doi.org/10.1016/S0042-6822(03)00242-3 |
||||
| 164 Paloheimo O, Ihalainen TO, Tauriainen S, Valilehto O, Kirjavainen S, Niskanen EA, Laakkonen JP, Hyoty H, Vihinen-Ranta M: Coxsackievirus B3-induced cellular protrusions: structural characteristics and functional competence. J Virol 2011;85:6714-6724. https://doi.org/10.1128/JVI.00247-10 |
||||
| 165 Agol VI, Belov GA, Bienz K, Egger D, Kolesnikova MS, Romanova LI, Sladkova LV, Tolskaya EA: Competing death programs in poliovirus-infected cells: commitment switch in the middle of the infectious cycle. J Virol 2000;74:5534-5541. https://doi.org/10.1128/JVI.74.12.5534-5541.2000 |
||||
| 166 Momoeda M, Wong S, Kawase M, Young NS, Kajigaya S: A putative nucleoside triphosphate-binding domain in the nonstructural protein of B19 parvovirus is required for cytotoxicity. J Virol 1994;68:8443-8446. https://doi.org/10.1128/jvi.68.12.8443-8446.1994 |
||||
| 167 Sol N, Le Junter J, Vassias I, Freyssinier JM, Thomas A, Prigent AF, Rudkin BB, Fichelson S, Morinet F: Possible interactions between the NS-1 protein and tumor necrosis factor alpha pathways in erythroid cell apoptosis induced by human parvovirus B19. J Virol 1999;73:8762-8770. https://doi.org/10.1128/JVI.73.10.8762-8770.1999 |
||||
| 168 Kytö V, Saraste A, Saukko P, Henn V, Pulkki K, Vuorinen T, Voipio-Pulkki LM: Apoptotic cardiomyocyte death in fatal myocarditis. Am J Cardiol 2004;94:746-750. https://doi.org/10.1016/j.amjcard.2004.05.056 |
||||
| 169 Clarke P, Tyler KL: Apoptosis in animal models of virus-induced disease. Nat Rev Microbiol 2009;7:144-155. https://doi.org/10.1038/nrmicro2071 |
||||
| 170 Wang YF, Wang XY, Ren Z, Qian CW, Li YC, Kaio K, Wang QD, Zhang Y, Zheng LY, Jiang JH, Yang CR, Liu Q, Zhang YJ: Phyllaemblicin B inhibits Coxsackie virus B3 induced apoptosis and myocarditis. Antiviral Res 2009;84:150-158. https://doi.org/10.1016/j.antiviral.2009.08.004 |
||||
| 171 Wang Q, Zhu Q, Ye Q, Wang J, Dong Q, Chen Y, Wang M, Fu Y, Wu R, Wu T: STAT3 Suppresses Cardiomyocytes Apoptosis in CVB3-Induced Myocarditis Via Survivin. Front Pharmacol 2020;11:613883. https://doi.org/10.3389/fphar.2020.613883 |
||||
| 172 Schulze K, Witzenbichler B, Christmann C, Schultheiss HP: Disturbance of myocardial energy metabolism in experimental virus myocarditis by antibodies against the adenine nucleotide translocator. Cardiovasc Res 1999;44:91-100. https://doi.org/10.1016/S0008-6363(99)00204-7 |
||||
| 173 Xu J, Nie HG, Zhang XD, Tian Y, Yu B: Down-regulated energy metabolism genes associated with mitochondria oxidative phosphorylation and fatty acid metabolism in viral cardiomyopathy mouse heart. Mol Biol Rep 2011;38:4007-4013. https://doi.org/10.1007/s11033-010-0519-y |
||||
| 174 Sin J, McIntyre L, Stotland A, Feuer R, Gottlieb RA: Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J Virol 2017;91:e01347-17. https://doi.org/10.1128/JVI.01347-17 |
||||
| 175 Lin L, Zhang M, Yan R, Shan H, Diao J, Wei J: Inhibition of Drp1 attenuates mitochondrial damage and myocardial injury in Coxsackievirus B3 induced myocarditis. Biochem Biophys Res Commun 2017;484:550-556. https://doi.org/10.1016/j.bbrc.2017.01.116 |
||||
| 176 Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ: Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 2003;278:36027-36031. https://doi.org/10.1074/jbc.M304854200 |
||||
| 177 Grivennikova VG, Vinogradov AD: Generation of superoxide by the mitochondrial Complex I. Biochim Biophys Acta 2006;1757:553-561. https://doi.org/10.1016/j.bbabio.2006.03.013 |
||||
| 178 Kannan K, Jain SK: Oxidative stress and apoptosis. Pathophysiology 2000;7:153-163. https://doi.org/10.1016/S0928-4680(00)00053-5 |
||||
| 179 Ebermann L, Wika S, Klumpe I, Hammer E, Klingel K, Lassner D, Volker U, Erben U, Zeichhardt H, Schultheiss HP, Dorner A: The mitochondrial respiratory chain has a critical role in the antiviral process in Coxsackievirus B3-induced myocarditis. Lab Invest 2012;92:125-134. https://doi.org/10.1038/labinvest.2011.145 |
||||
| 180 Xie B, Zhou JF, Lu Q, Li CJ, Chen P: Oxidative stress in patients with acute coxsackie virus myocarditis. Biomed Environ Sci 2002;15:48-57. | ||||
| 181 Peischard S, Ho HT, Piccini I, Strutz-Seebohm N, Röpke A, Liashkovich I, Gosain H, Rieger B, Klingel K, Eggers B, Marcus K, Linke WA, Müller FU, Ludwig S, Greber B, Busch K, Seebohm G: The first versatile human iPSC-based model of ectopic virus induction allows new insights in RNA-virus disease. Sci Rep 2020;10:16804. https://doi.org/10.1038/s41598-020-72966-9 |
||||
| 182 Chi J, Yu S, Liu C, Zhao X, Zhong J, Liang Y, Ta N, Yin X, Zhao D: Nox4-dependent ROS production is involved in CVB. Biochem Biophys Res Commun 2018;503:1641-1644. https://doi.org/10.1016/j.bbrc.2018.07.093 |
||||
| 183 Eisner DA, Caldwell JL, Kistamás K, Trafford AW: Calcium and Excitation-Contraction Coupling in the Heart. Circ Res 2017;121:181-195. https://doi.org/10.1161/CIRCRESAHA.117.310230 |
||||
| 184 Priest BT, McDermott JS: Cardiac ion channels. Channels (Austin) 2015;9:352-359. https://doi.org/10.1080/19336950.2015.1076597 |
||||
| 185 Salerno F, Girerd N, Chalabreysse L, Billaud G, Lina B, Chevalier P: Myocarditis and cardiac channelopathies: a deadly association? Int J Cardiol 2011;147:468-470. https://doi.org/10.1016/j.ijcard.2011.01.019 |
||||
| 186 Steinke K, Sachse F, Ettischer N, Strutz-Seebohm N, Henrion U, Rohrbeck M, Klosowski R, Wolters D, Brunner S, Franz WM, Pott L, Munoz C, Kandolf R, Schulze-Bahr E, Lang F, Klingel K, Seebohm G: Coxsackievirus B3 modulates cardiac ion channels. FASEB J 2013;27:4108-4121. https://doi.org/10.1096/fj.13-230193 |
||||
| 187 Seebohm G, Strutz-Seebohm N, Ureche ON, Henrion U, Baltaev R, Mack AF, Korniychuk G, Steinke K, Tapken D, Pfeufer A, Kääb S, Bucci C, Attali B, Merot J, Tavare JM, Hoppe UC, Sanguinetti MC, Lang F: Long QT syndrome-associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ Res 2008;103:1451-1457. https://doi.org/10.1161/CIRCRESAHA.108.177360 |
||||
| 188 Ilnytska O, Santiana M, Hsu NY, Du WL, Chen YH, Viktorova EG, Belov G, Brinker A, Storch J, Moore C, Dixon JL, Altan-Bonnet N: Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013;14:281-293. https://doi.org/10.1016/j.chom.2013.08.002 |
||||
| 189 Anacker C, Cattaneo A, Musaelyan K, Zunszain PA, Horowitz M, Molteni R, Luoni A, Calabrese F, Tansey K, Gennarelli M, Thuret S, Price J, Uher R, Riva MA, Pariante CM: Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci U S A 2013;110:8708-8713. https://doi.org/10.1073/pnas.1300886110 |
||||
| 190 Su YG, Yang YZ, Bao WS, Liu GX, Ge JB, Chen H: Effects of taurine and Astragalus membranaceus on ion currents and their expression in cardiomyocytes after CVB3 infection. Drug Development Research 2003;58:57-60. https://doi.org/10.1002/ddr.10130 |
||||
| 191 Cheng J, Wen J, Wang N, Wang C, Xu Q, Yang Y: Ion Channels and Vascular Diseases. Arterioscler Thromb Vasc Biol 2019;39:e146-e156. https://doi.org/10.1161/ATVBAHA.119.312004 |
||||
| 192 Dorsch S, Liebisch G, Kaufmann B, von Landenberg P, Hoffmann JH, Drobnik W, Modrow S: The VP1 unique region of parvovirus B19 and its constituent phospholipase A2-like activity. J Virol 2002;76:2014-2018. https://doi.org/10.1128/JVI.76.4.2014-2018.2002 |
||||
| 193 Hernández-Araiza I, Morales-Lázaro SL, Canul-Sánchez JA, Islas LD, Rosenbaum T: Role of lysophosphatidic acid in ion channel function and disease. J Neurophysiol 2018;120:1198-1211. https://doi.org/10.1152/jn.00226.2018 |
||||
| 194 Law SH, Chan ML, Marathe GK, Parveen F, Chen CH, Ke LY: An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int J Mol Sci 2019;20:1149. https://doi.org/10.3390/ijms20051149 |
||||
| 195 Lupescu A, Bock CT, Lang PA, Aberle S, Kaiser H, Kandolf R, Lang F: Phospholipase A2 activity-dependent stimulation of Ca2+ entry by human parvovirus B19 capsid protein VP1. J Virol 2006;80:11370-11380. https://doi.org/10.1128/JVI.01041-06 |
||||
| 196 Behringer EJ: Calcium and electrical signaling in arterial endothelial tubes: New insights into cellular physiology and cardiovascular function. Microcirculation 2017; DOI: 10.1111/micc.12328. https://doi.org/10.1111/micc.12328 |
||||
| 197 Ahmed M, Honisch S, Pelzl L, Fezai M, Hosseinzadeh Z, Bock CT, Kandolf R, Lang F: Up-regulation of epithelial Na(+) channel ENaC by human parvovirus B19 capsid protein VP1. Biochem Biophys Res Commun 2015;468:179-184. https://doi.org/10.1016/j.bbrc.2015.10.137 |
||||
| 198 Ahmed M, Almilaji A, Munoz C, Elvira B, Shumilina E, Bock CT, Kandolf R, Lang F: Down-regulation of K⁺ channels by human parvovirus B19 capsid protein VP1. Biochem Biophys Res Commun 2014;450:1396-1401. https://doi.org/10.1016/j.bbrc.2014.07.003 |
||||
| 199 Ahmed M, Elvira B, Almilaji A, Bock CT, Kandolf R, Lang F: Down-regulation of inwardly rectifying Kir2.1 K+ channels by human parvovirus B19 capsid protein VP1. J Membr Biol 2015;248:223-229. https://doi.org/10.1007/s00232-014-9762-9 |
||||
| 200 Zádori Z, Szelei J, Lacoste MC, Li Y, Gariépy S, Raymond P, Allaire M, Nabi IR, Tijssen P: A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell 2001;1:291-302. https://doi.org/10.1016/S1534-5807(01)00031-4 |
||||
| 201 Ho HT, Peischard S, Strutz-Seebohm N, Klingel K, Seebohm G: Myocardial Damage by SARS-CoV-2: Emerging Mechanisms and Therapies. Viruses 2021;13:1880. https://doi.org/10.3390/v13091880 |
||||
| 202 Kraft L, Sauter M, Seebohm G, Klingel K: In vitro Model Systems of Coxsackievirus B3-Induced Myocarditis: Comparison of Commonly Used Cell Lines and Characterization of CVB3-Infected iCell. Viruses 2021;13:1835. https://doi.org/10.3390/v13091835 |
||||