×
![]()
Corresponding Author: Cenk Suphioglu
NeuroAllergy Research Laboratory (NARL), School of Life and Environmental Sciences (LES), Faculty of Science, Engineering and Built Environment (SEBE), Deakin University,
75 Pigdons Road, Waurn Ponds, Victoria 3216 (Australia)
E-Mail cenk.suphioglu@deakin.edu.au
Apolipoprotein E4 as a Novel Treatment Target for Alzheimer’s Disease
Jamieson Lachlan Jervies Samuel Peter King Cenk Suphioglu
NeuroAllergy Research Laboratory (NARL), School of Life and Environmental Sciences (LES), Faculty of Science, Engineering and Built Environment (SEBE), Deakin University, Waurn Ponds, Victoria, Australia,
Neurodegenerative Diseases
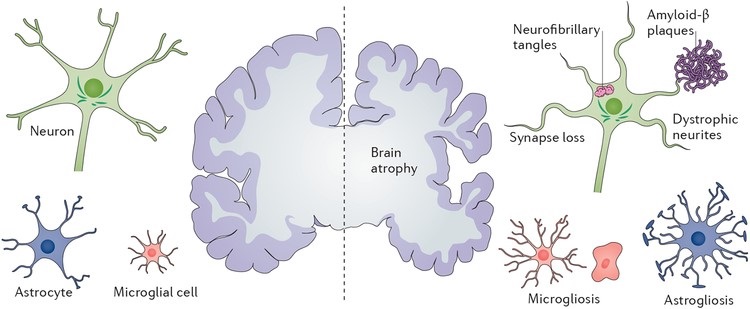
Neurodegenerative diseases present in a variety of conditions, however, are caused by the damage, death and loss of neurons. The degradation of these neuronal cells causes many debilitating and often incurable diseases which leads to a large burden not only on the affected individual but their support network and society both personally and financially. The impact of disease forms a progressive decline in cognitive and motor function which at later stages of progression requires extensive care and assistance [1].
Neurodegenerative diseases are becoming more prevalent worldwide due to current increases in life expectancy and therefore ageing population. The increase in the ageing population is causing disease rates to increase as age is the primary risk factor for most neurodegenerative diseases with one out of every ten individuals aged over 65 years impacted by Alzheimer’s disease alone [2].
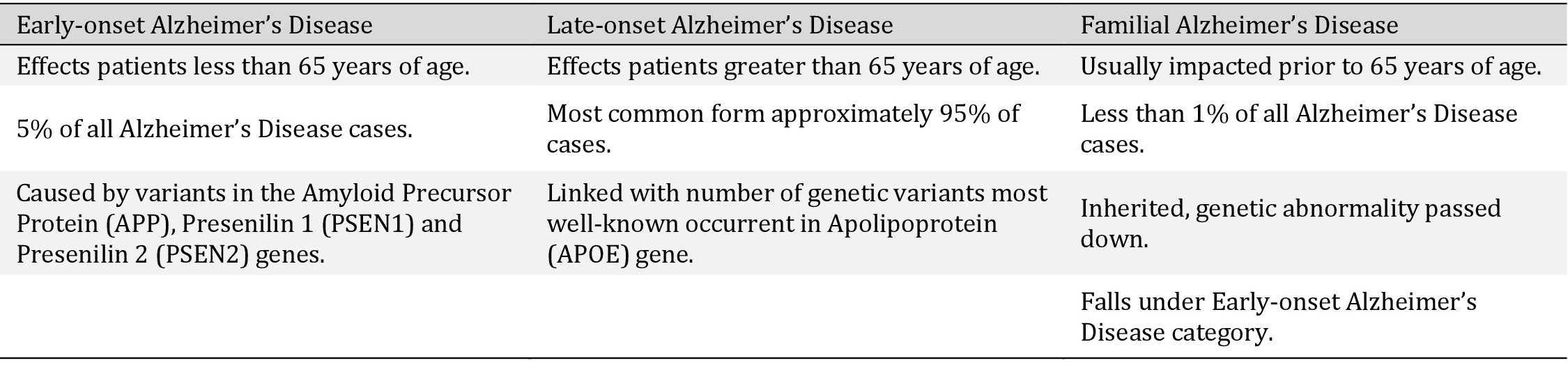
Representing the key differences between the 3 types of Alzheimer’s Disease. Showing the difference in those affected, occurrence rates and main genetic risk factors from each category
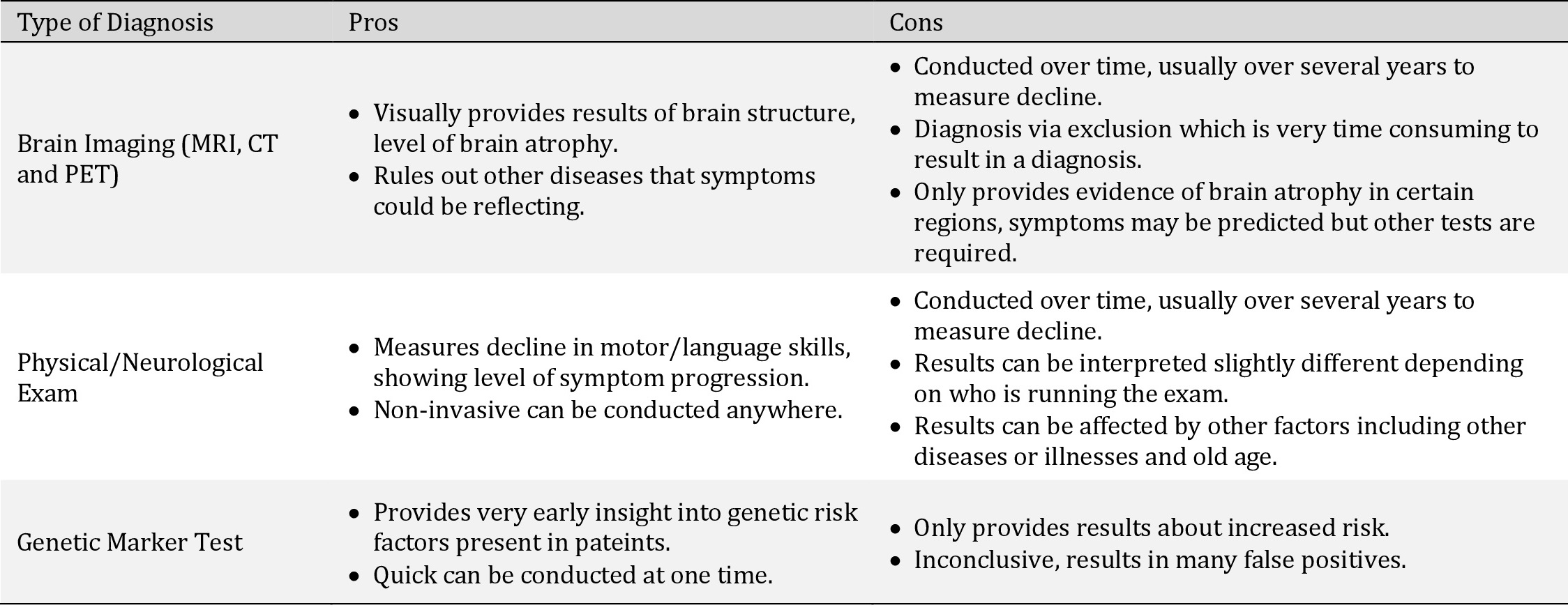
Pros and cons of various methods of diagnosis. Generally, these methods are used in conjunction to determine Alzheimer’s Disease diagnosis as one test alone is inconclusive
The authors declare that no conflicts of interest exist.
| 1 Katsuno M, Sahashi K, Iguchi Y, Hashizume A: Preclinical progression of neurodegenerative diseases. Nagoya J Med Sci 2018;80:289-298. | ||||
| 2 Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA: Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 2019;15:565-581. https://doi.org/10.1038/s41582-019-0244-7 |
||||
| 3 Weller J, Budson A: Current understanding of Alzheimer's disease diagnosis and treatment. F1000Res 2018;7:1161. https://doi.org/10.12688/f1000research.14506.1 |
||||
| 4 Australien Institute of Health and Welfare: Causes of death (leading causes of death 2018). Release Date 23 Jul 2020. URL: https://www.aihw.gov.au/reports/australias-health/causes-of-death. | ||||
| 5 Awada AA: Early and late-onset Alzheimer's disease: What are the differences?. J Neurosci Rural Pract 2015;6:455-456. https://doi.org/10.4103/0976-3147.154581 |
||||
| 6 Bondi MW, Edmonds EC, Salmon DP: Alzheimer's Disease: Past, Present, and Future. J Int Neuropsychol Soc 2017;23:818-831. https://doi.org/10.1017/S135561771700100X |
||||
| 7 Chi H, Chang HY, Sang TK: Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int J Mol Sci 2018;19:3082. https://doi.org/10.3390/ijms19103082 |
||||
| 8 Raji CA, Lopez OL, Kuller LH, Carmichael OT, Becker JT: Age, Alzheimer disease, and brain structure. Neurology 2009;73:1899-1905. https://doi.org/10.1212/WNL.0b013e3181c3f293 |
||||
| 9 Congdon EE, Sigurdsson, EM: Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 2018;14:399-415. https://doi.org/10.1038/s41582-018-0013-z |
||||
| 10 Buchman AS, Bennett DA: Loss of motor function in preclinical Alzheimer's disease. Expert Rev Neurother 2011;11:665-676. https://doi.org/10.1586/ern.11.57 |
||||
| 11 Bu XL, Jiao SS, Lian Y, Wang YJ: Perspectives on the Tertiary Prevention Strategy for Alzheimer's Disease. Curr Alzheimer Res 2016;13:307-316. https://doi.org/10.2174/1567205013666151215110114 |
||||
| 12 Chen G, Xu T, Yan Y, Zhou Y, Jiang Y, Melcher K, Xu HE: Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 2017;38:1205-1235. https://doi.org/10.1038/aps.2017.28 |
||||
| 13 Wang S, Mims PN, Roman RJ, Fan F: Is Beta-Amyloid Accumulation a Cause or Consequence of Alzheimer's Disease?. J Alzheimers Parkinsonism Dement 2016:1:7. | ||||
| 14 Seeman P, Seeman N: Alzheimer's Disease: β-Amyloid Plaque Formation in Human Brain. Synapse 2011;65:1289-1297. https://doi.org/10.1002/syn.20957 |
||||
| 15 Takahashi R, Nagao T, Gouras G: Plaque Formation and the Intraneuronal Accumulation of β-Amyloid in Alzheimer's Disease. Pathol Int 2017;67:185-193. https://doi.org/10.1111/pin.12520 |
||||
| 16 Guo T, Noble W, Hanger DP: Roles of tau protein in health and disease. Acta Neuropathol 2017;133:665-704. https://doi.org/10.1007/s00401-017-1707-9 |
||||
| 17 Ittner A, Ittner LM: Dendritic Tau in Alzheimer's Disease. Neuron 2018;99:13-27. https://doi.org/10.1016/j.neuron.2018.06.003 |
||||
| 18 Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G: Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. Int J Mol Sci 2014;15:4671-4713. https://doi.org/10.3390/ijms15034671 |
||||
| 19 Biswas S, Kalil K: The Microtubule-Associated Protein Tau Mediates the Organization of Microtubules and Their Dynamic Exploration of Actin-Rich Lamellipodia and Filopodia of Cortical Growth Cones. J Neurosci 2018;38:291-307. https://doi.org/10.1523/JNEUROSCI.2281-17.2017 |
||||
| 20 Alonso AD, Cohen LS, Corbo C, Morozova V, ElIdrissi A, Phillips G, Kleiman FE: Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front Cell Neurosci 2018;12:338. https://doi.org/10.3389/fncel.2018.00338 |
||||
| 21 Noble W, Hanger DP, Miller CCJ, Lovestone S: The Importance of Tau Phosphorylation for Neurodegenerative Diseases. Front Neurol 2013;4:83. https://doi.org/10.3389/fneur.2013.00083 |
||||
| 22 Wang Y, Mandelkow E: Tau in physiology and pathology. Nat Rev Neurosci 2016;17:22-35. https://doi.org/10.1038/nrn.2015.1 |
||||
| 23 Luppi M, Hitrec T, Di Cristoforo A, Squarcio F, Stanzani A, Occhinegro A, Chiavetta P, Tupone D, Zamboni G, Amici R, Cerri M: Phosphorylation and Dephosphorylation of Tau Protein During Synthetic Torpor. Front Neuroanat 2019;13:57. https://doi.org/10.3389/fnana.2019.00057 |
||||
| 24 Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, Rall SC: Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. J Biol Chem 1989;264:9094-9101. https://doi.org/10.1016/S0021-9258(18)81907-X |
||||
| 25 Kim J, Jiang H, Park S, Eltorai AEM, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM: Haploinsufficiency of Human APOE Reduces Amyloid Deposition in a Mouse Model of Amyloid-β Amyloidosis. J Neurosci 2011;31:18007-18012. https://doi.org/10.1523/JNEUROSCI.3773-11.2011 |
||||
| 26 Zhao L, Wu L: ApoE2 and Alzheimer′s disease: time to take a closer look. Neural Regen Res 2016;11:412. https://doi.org/10.4103/1673-5374.179044 |
||||
| 27 Huang YWA, Zhou B, Wernig M, Südhof TC: ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017;168:427-441. https://doi.org/10.1016/j.cell.2016.12.044 |
||||
| 28 Fernandez CG, Hamby ME, McReynolds ML, Ray WJ: The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer's Disease. Front Aging Neurosci 2019;11:14. https://doi.org/10.3389/fnagi.2019.00014 |
||||
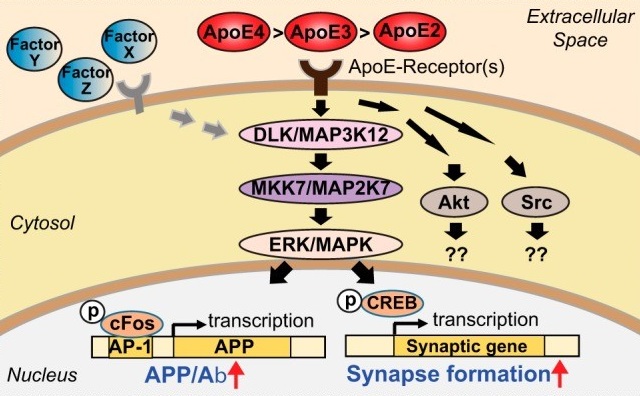
| 29 Huang YWA, Zhou B, Nabet AM, Wernig M, Südhof TC: Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer's Disease Risk. J Neurosci 2019;39:7408-7427. https://doi.org/10.1523/JNEUROSCI.2994-18.2019 |
||||
| 30 Hemonnot A-L, Hua J, Ulmann L, Hirbec H: Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front Aging Neurosci 2019;11:233. https://doi.org/10.3389/fnagi.2019.00233 |
||||
| 31 Ries M, Sastre M: Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front Aging Neurosci 2016;8:160. https://doi.org/10.3389/fnagi.2016.00160 |
||||
| 32 Area-Gomez E, Larrea D, Pera M, Agrawal RR, Guilfoyle DN, Pirhaji L, Shannon K, Arain HA, Ashok A, Chen Q, Dillman AA, Figueroa HY, Cookson MR, Gross SS, Fraenkel E, Duff KE, Nuriel T: APOE4 is Associated with Differential Regional Vulnerability to Bioenergetic Deficits in Aged APOE Mice. Sci Rep 2020;10:1-20. https://doi.org/10.1038/s41598-020-61142-8 |
||||
| 33 Kang SS, Ahn EH, Liu X, Bryson M, Miller GW, Weinshenke, D, Ye K: ApoE4 inhibition of VMAT2 in the locus coeruleus exacerbates Tau pathology in Alzheimer's disease. Acta Neuropathol 2021;142:139-158. https://doi.org/10.1007/s00401-021-02315-1 |
||||
| 34 Kang SS, Ahn EH, Ye K: Delta-secretase cleavage of Tau mediates its pathology and propagation in Alzheimer's disease. Exp Mol Med 2020;52:1275-1287. https://doi.org/10.1038/s12276-020-00494-7 |
||||
| 35 Lemprière S: APOE4 provokes tau aggregation via inhibition of noradrenaline transport. Nat Rev Neurol 2021;17:328-328. https://doi.org/10.1038/s41582-021-00511-x |
||||
| 36 Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, Melzer D: APOE e4 genotype predicts severe COVID-19 in the UK Biobank community cohort. J Gerontol A Biol Sci Med Sci 2020;75:2231-2232. https://doi.org/10.1093/gerona/glaa131 |
||||
| 37 Wang C, Zhang M, Garcia G, Tian E, Cui Q, Chen X, Sun G, Wang J, Arumugaswami V, Shi Y: ApoE-Isoform-Dependent SARS-CoV-2 Neurotropism and Cellular Response. Cell Stem Cell 2021;28:331-342. https://doi.org/10.1016/j.stem.2020.12.018 |
||||
| 38 Xiong N, Schiller MR, Li J, Chen X, Lin Z: Severe COVID-19 in Alzheimer's disease: APOE4's fault again?. Alzheimers Res Ther 2021;13:111. https://doi.org/10.1186/s13195-021-00858-9 |
||||
| 39 Hippius H, Neundörfer G: The discovery of Alzheimer's disease. Dialogues Clin Neurosci 2003;5:101-108. https://doi.org/10.31887/DCNS.2003.5.1/hhippius |
||||
| 40 Ryan NS, Rossor MN, Fox NC: Alzheimer's disease in the 100 years since Alzheimer's death. Brain 2015;138:3816-3821. https://doi.org/10.1093/brain/awv316 |
||||
| 41 DeTure MA, Dickson DW: The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener 2019;14:32. https://doi.org/10.1186/s13024-019-0333-5 |
||||
| 42 Zetterberg H, Burnham SC: Blood-based molecular biomarkers for Alzheimer's disease. Mol Brain 2019;12:26. https://doi.org/10.1186/s13041-019-0448-1 |
||||
| 43 Park M, Moon WJ: Structural MR Imaging in the Diagnosis of Alzheimer's Disease and Other Neurodegenerative Dementia: Current Imaging Approach and Future Perspectives. Korean J Radiol 2016;17:827-845. https://doi.org/10.3348/kjr.2016.17.6.827 |
||||
| 44 Suppiah S, Didier MA, Vinjamuri S: The Who, When, Why, and How of PET Amyloid Imaging in Management of Alzheimer's Disease-Review of Literature and Interesting Images. Diagnostics (Basel) 2019;9:65. https://doi.org/10.3390/diagnostics9020065 |
||||
| 45 Khanahmadi M, Farhud DD, Malmir M: Genetic of Alzheimer's Disease: A Narrative Review Article. Iran J Public Health 2015;44:892-901. | ||||
| 46 Lanoiselée HM, Nicolas G, Wallon D, Rovelet-Lecrux A, Lacour M, Rousseau S, Richard AC, Pasquier F, Rollin-Sillaire A, Martinaud O, Quillard-Muraine M, de la Sayette V, Boutoleau-Bretonniere C, Etcharry-Bouyx F, Chauviré V, Sarazin M, le Ber I, Epelbaum S, Jonveaux T, Rouaud O: APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 2017;14:e1002270. https://doi.org/10.1371/journal.pmed.1002270 |
||||
| 47 Delabio R, Rasmussen L, Mizumoto I, Viani GA, Chen E, Villares J, Costa IB, Turecki G, Linde SA, Smith MC, Payão SL: PSEN1 and PSEN2 gene expression in Alzheimer's disease brain: a new approach. J Alzheimers Dis 2014;42:757-760. https://doi.org/10.3233/JAD-140033 |
||||
| 48 Yiannopoulou KG, Papageorgiou SG: Current and Future Treatments in Alzheimer Disease: An Update. J Cent Nerv Syst Dis 2020; DOI: 10.1177/1179573520907397. https://doi.org/10.1177/1179573520907397 |
||||
| 49 Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G: Therapies for Prevention and Treatment of Alzheimer's Disease. BioMed Res Int 2016; DOI: 10.1155/2016/2589276. https://doi.org/10.1155/2016/2589276 |
||||
| 50 Picciotto MR, Higley MJ, Mineur YS: Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012;76:116-129. https://doi.org/10.1016/j.neuron.2012.08.036 |
||||
| 51 Eldufani J, Blaise G: The role of acetylcholinesterase inhibitors such as neostigmine and rivastigmine on chronic pain and cognitive function in aging: A review of recent clinical applications. Alzheimers Dement 2019;5:175-183. https://doi.org/10.1016/j.trci.2019.03.004 |
||||
| 52 Liu J, Chang L, Song Y, Li H, Wu Y: The Role of NMDA Receptors in Alzheimer's Disease. Front Neurosci 2019;13:43. https://doi.org/10.3389/fnins.2019.00043 |
||||
| 53 Wang R, Reddy PH: Role of Glutamate and NMDA Receptors in Alzheimer's Disease. J Alzheimers Dis 2017;57:1041-1048. https://doi.org/10.3233/JAD-160763 |
||||
| 54 Atri A: Current and Future Treatments in Alzheimer's Disease. Semin Neurol 2019;39:227-240. https://doi.org/10.1055/s-0039-1678581 |
||||
| 55 Nuriel T, Angulo SL, Khan U, Ashok A, Chen Q, Figueroa HY, Emrani S, Liu L, Herman M, Barrett G, Savage V, Buitrago L, Cepeda-Prado E, Fung C, Goldberg E, Gross SS, Hussaini SA, Moreno H, Small SA, Duff KE: Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer's disease-like pathology. Nat Commun 2017;8:1464. https://doi.org/10.1038/s41467-017-01444-0 |
||||
| 56 Sharman MJ, Morici M, Hone E, Berger T, Taddei K, Martins IJ, Lim WLF, Singh S, Wenk MR, Ghiso J, Buxbaum JD, Gandy S, Martins RN: APOE genotype results in differential effects on the peripheral clearance of amyloid-beta42 in APOE knock-in and knock-out mice. J Alzheimers Dis 2010;21:403-409. https://doi.org/10.3233/JAD-2010-100141 |
||||
| 57 Li Z: New APOE-related therapeutic options for Alzheimer's disease. AIP Conf Proc 2019; DOI: 10.1063/1.5085515. https://doi.org/10.1063/1.5085515 |
||||
| 58 Chou CY, Jen WP, Hsieh YH, Shiao MS, Chang GG: Structural and Functional Variations in Human Apolipoprotein E3 and E4. J Biol Chem 2006;281;13333-13344. https://doi.org/10.1074/jbc.M511077200 |
||||
| 59 Wang M, Turko IV: Mass spectrometry quantification revealed accumulation of C-terminal fragment of apolipoprotein E in the Alzheimer's frontal cortex. PloS One 2013;8:e61498. https://doi.org/10.1371/journal.pone.0061498 |
||||