×
![]()
Corresponding Author: Peter Boor
Institute of Pathology, University Hospital Aachen, RWTH Aachen University, Pauwelsstr. 30, 52057 Aachen (Germany)
Tel. +49 241 80 35101, E-Mail pboor@ukaachen.de
A Hypercaloric Diet Induces Early Podocyte Damage in Aged, Non-Diabetic Rats
Claudia Seikrita,b Eva Lausbergb,c Eva Miriam Buhlb,d Robert Gaspare Tamas Tabif Marija Hefferg Eszter Duczah Anita Sztojkov-Ivanovh Adrienn B. Seresh Kalman Szucse Vedrana Ivicg Jürgen Floegea Sandor G. Varii Peter Boora,b,d Barbara Mara Klinkhammerb
aDivision of Nephrology and Clinical Immunology, RWTH Aachen University, Aachen, Germany, bInstitute of Pathology, University Hospital Aachen, RWTH Aachen University, Aachen, Germany, cInstitute of Human Genetics, RWTH Aachen University, Aachen, Germany, dElectron Microscopy Facility, RWTH Aachen University Hospital, Aachen, Germany, eDepartment of Pharmacology and Pharmacotherapy, Faculty of Medicine, Interdisciplinary Excellence Centre, University of Szeged, Szeged, Hungary, fDepartment of Pharmacodynamics, Faculty of Pharmacy, Semmelweis University, Budapest, Hungary, gDepartment of Medical Biology and Genetics, Faculty of Medicine, J. J. Strossmayer University of Osijek, Osijek, Croatia, hDepartment of Pharmacodynamics and Biopharmacy, University of Szeged, Szeged, Hungary, iCedars-Sinai Medical Center, International Research and Innovation in Medicine Program, Los Angeles, CA, USA
Introduction
The prevalence of obesity, metabolic syndrome, and chronic kidney disease (CKD) is increasing worldwide [1]. Obesity and CKD are interdependent, as obesity is a known risk factor for CKD [2-5]. Especially central or visceral obesity has been linked to unfavorable outcomes and the development of CKD [6]. Development of insulin resistance and glomerular hyperfiltration are thought to contribute to the pathophysiological mechanisms aggravating CKD in obese individuals [7-10]. But even without clinically apparent glucose intolerance, glomeruli of obese patients show mild diabetes-like changes characterized by mesangial matrix expansion, thickening of the glomerular basement membrane (GBM), and glomerular hypertrophy, potentially leading to focal segmental glomerulosclerosis (FSGS), a hallmark of CKD [5, 11, 12]. Podocyte injury is the initiating step in the development of FSGS. In obesity glomerular hyperfiltration is assumed to induce thickening of the GBM and an increase of the glomerular size provoking podocyte hypertrophy as a response to the increased filtration area. This leads to podocyte shear stress, apoptosis, and potentially podocyte detachment resulting in increasing numbers of glomeruli presenting with FSGS. In patients, these pathological changes lead to proteinuria as a consequence of impaired filtration function, and to a progressive loss of kidney function. FSGS in obesity-related glomerulopathy occurs often in middle-aged patients taking a slowly progressing clinical course over time [11]. Of note, most preclinical studies use young animals. The metabolic syndrome was mostly studied in young and often genetically modified animals, which does not reflect the clinical reality of most patients. In general, the consequences of the metabolic syndrome do not manifest before older age.
Several drugs are being used for the treatment of patients with diabetes or metabolic syndrome. Two of these drugs are the glucagon-like peptide receptor 1 agonist (GLP-1RA) liraglutide and the biguanide derivate metformin. GLP-1RAs, such as liraglutide or semaglutide, were shown to be effective in the treatment of diabetes mellitus in large clinical trials, e.g. the LEADER trial [13, 14], and liraglutide has been shown to slow down the incidence of diabetes in obese patients [15]. GLP-1RAs improve glycemic control and promote weight loss and have subsequently been approved also for the treatment of obesity by the FDA [16]. Renal benefits of the treatment have been reported in the LEADER and SUSTAIN 6 trials [13, 17, 18]. Several experimental studies showed a beneficial effect for GLP-1RAs in models of diabetic nephropathy, which included reduced tubulointerstitial renal damage, lowered expression of tubular injury markers, and attenuated renal accumulation of macrophages and T cells [19-21]. Renal benefits of liraglutide on obesity alone have been studied less well, yet giving some promising results [22, 23].
Metformin, a biguanide derivate, is the standard of care for patients with type 2 diabetes mellitus especially with concomitant obesity [24]. Metformin decreases hepatic gluconeogenesis and increases glucose uptake in muscles and peripheral tissue and thus lowers serum glucose levels and insulin secretion. In diverse studies, metformin has been shown to have a positive effect on weight loss in obese patients without the simultaneous onset of diabetes mellitus [25-27]. In models of diabetic nephropathy, metformin exerted renoprotective actions, e.g., by counteracting interstitial fibrosis or reducing podocyte damage and apoptosis [28-31].
Here we investigate the potential renal effects of a long-term high caloric high-fat-high-carbohydrate diet in aged rats. We focused on glomerular and podocyte injury as crucial and initiating pathological processes in obesity and metabolic syndrome-related renal diseases. We also analyzed potential effects of metformin or liraglutide treatment in these rats.
Materials and Methods
Animal model and experimental design
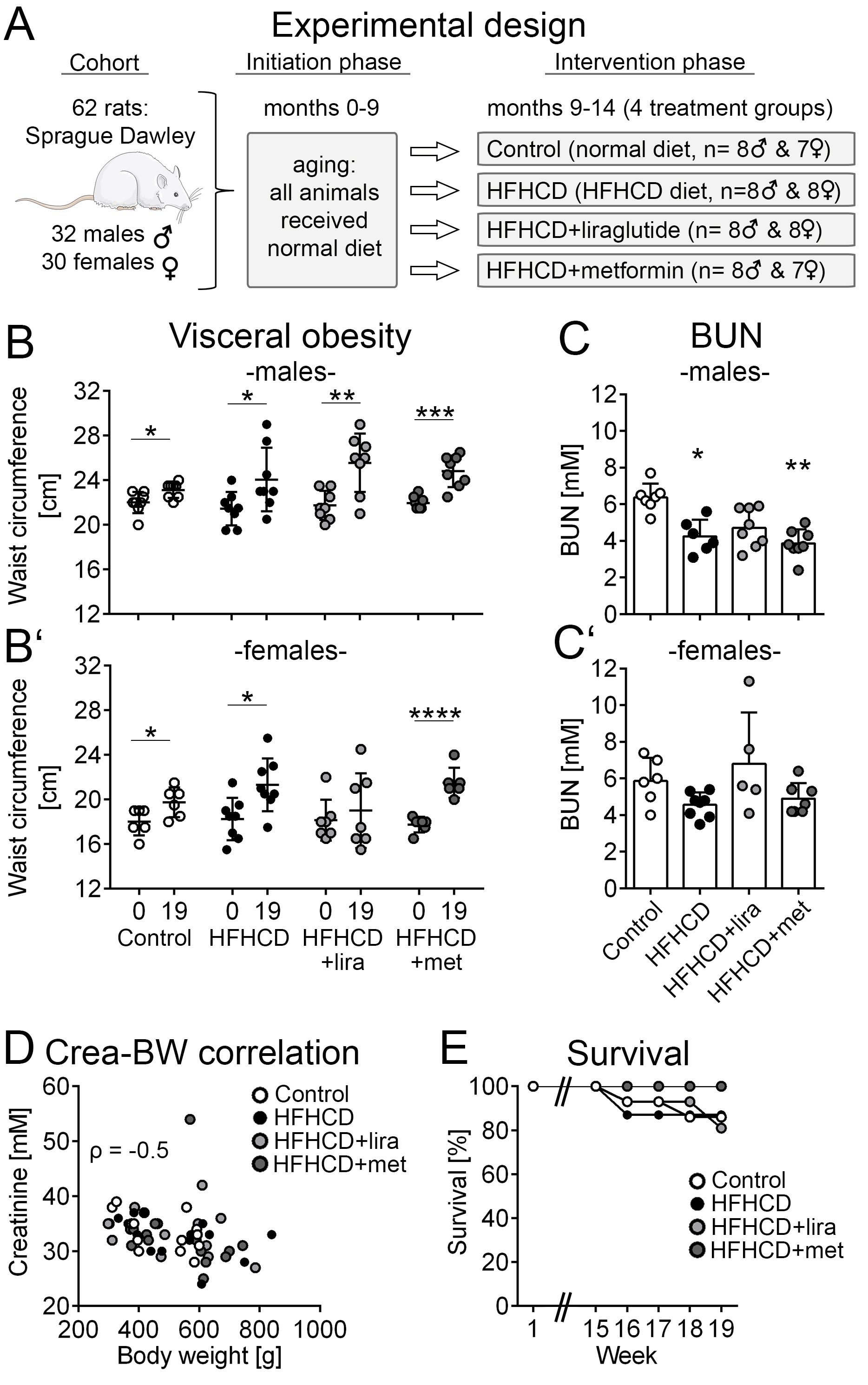
Sixty-two Sprague Dawley rats (32 male and 30 female) aged 3 weeks were obtained from Charles River Ltd (Charles River, Sulzheim, Germany). They were fed with a normal chow (Altromin 1324, Altromin GmbH, Lage, Germany) diet during an initial phase of 9 months. Afterwards, they were randomized into four different groups. The control group (8 males and 7 females) was maintained on a normal diet and compared to three intervention groups. The HFHCD group (8 males and 8 females) was fed with a high-fat-high-carbohydrate diet (Altromin C 1011, Altromin GmbH, Lage, Germany). In the other two intervention groups, animals on HFHCD received either 0.3 mg/kg/d liraglutide subcutaneously (HFHCD+liraglutide group, 8 males and 8 females) or 50 mg/kg/d metformin (HFHCD+metformin group, 8 males and 7 females). During this intervention phase (months 9 to 14), 2 animals (1 male, 1 female) died in the control group, while 2 died in the HFHCD group (2 males). The HFHCD+liraglutide group lost 3 animals (3 females). In the HFHCD+metformin group no animal died. All surviving animals were sacrificed after 19 weeks of treatment at an age of 14 months (Fig. 1A). Kidneys were collected and processed for further analyses. Except for electron microscopy, all available specimens were included in all analyses.
Histology and immunohistochemistry
Kidney tissue was fixed in methyl Carnoy’s fixative, then dehydrated and embedded in paraffin. 1 µm sections (longitudinal kidney cross-sections) were cut and paraffin was removed by washing slides in xylene for 5 minutes (3 times), and then rehydrated in decreasing concentrations of ethanol. Slides were stained with periodic acid-Schiff (PAS) and counterstained with hematoxylin for routine morphological evaluation of glomerular and tubulointerstitial renal damage.
Gömöri-silver-staining was performed for evaluation of FSGS. In short, slides were placed in 0.5% thiosemicarbazide for 5 minutes. After washing in distilled water for 5 minutes and heating in 65°C hot distilled water, they were incubated with methenamine silver solution for 10 minutes at 37°C and then for 10 minutes at 65°C. After a washing step at 65°C the slides were again incubated in new methenamine silver solution at 65°C for 20 minutes, washed, and placed in gold chloride solution for 15 minutes. Then another washing step was followed by 1 minute in 2% nitric acid formalin. They were counterstained with hematoxylin and eosin for 3-5 minutes.
For immunohistochemistry, after deparaffinization in xylene and rehydration in ethanol, endogenous peroxidase activity was blocked with 3% H2O2 in distilled water for 10 minutes at room temperature. Primary antibodies were applied for 1 hour in a humid chamber. After washing with PBS, biotinylated secondary antibodies were incubated for 30 minutes. PBS or primary antibody species IgG were used as a negative control. DAB (3,3’-diaminobenzidine) was used as horseradish peroxidase (HRP) substrate and methyl green was used for counterstaining. Slides were mounted with Histokitt (Carl Roth, Karlsruhe, Germany). All antibodies used are listed in Table 1.
Stained tissue slides were scanned using NanoZoomer 2.0 digital slide scanner (Hamamatsu, Herrsching am Ammersee, Germany). For computer-assisted quantification of glomerular size and immunostainings, all glomerular cross-sections per slide were photographed (original magnification x200) and further analyzed using the ImageJ software V1.48 (National Institutes of Health, Bethesda, MD), corresponding to 300-400 glomeruli per animal. The tuft size was analyzed in PAS stainings by measuring the area of tuft cross-sections by manual annotation. The positively stained glomerular area in desmin, nestin and podocin immunostainings was also analyzed by quantification in manual annotations of all glomerular tuft cross-sections per slide. In CD44 stainings, the numbers of glomeruli with positively stained parietal epithelial cells (PECs) were counted. For computer-based morphometric evaluation of the tubulointerstitium in collagen immunostainings, 20 photomicrographs (original magnification x100) were randomly taken per kidney cortex. Area fractions of positive staining were evaluated by computer-based morphometry using ImageJ. In lipocalin-2 (LCN2, also called neutrophil gelatinase-associated lipocalin (NGAL)) stainings the number of positively stained tubules was counted and in CD68 stainings the number of positively stained interstitial cells. Both results were normalized to the kidney cross-sectional area. In PAS stainings the tubulointerstitial injury was estimated as percent of damaged area, considering tubular cell flattening and loss of brush borders as well as tubular dilation and atrophy and tubulointerstitial inflammation and fibrosis. All analyses were performed in a blinded manner.
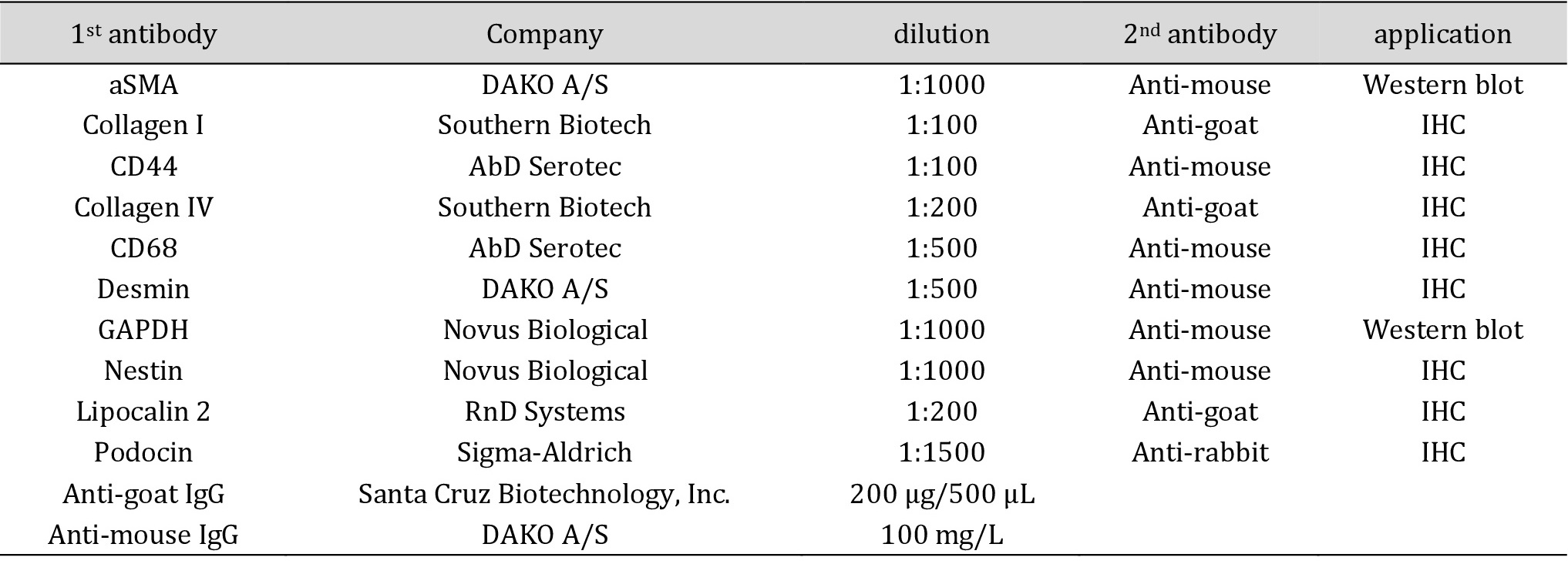
Antibodies for immunohistochemical staining and western blotting. aSMA = alpha smooth muscle actin, CD = cluster of differentiation, GAPDH = glyceraldehyde 3-phosphate dehydrogenase, IgG = immunoglobulin G, IHC = immunohistochemistry
mRNA tissue extraction
Kidney cortex was homogenized in lysis buffer using micro iron balls and RNA extraction was performed according to the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using random primers. Briefly, 1 µg RNA was added to a total volume of 20 µl and incubated 10 minutes at 75°C in the thermocycler (Applied Biosystems, Weiterstadt, Germany). After a short centrifugation, 10.2 µl master mix (a 30 μl reaction mix including 1 μg of total RNA, 1 μl of random primer (6 nt, 250 ng/μl; Roche, Basel, Switzerland), 6 μl of M-MLV reverse transcriptase buffer (Invitrogen, Carlsbad, CA), 1.5 μl of dNTP-mix (10 mM each; Carl Roth, Karlsruhe, Germany), 0.7 μl of RNase-inhibitor (40 U/μl, Promega, Madison, WI), 1 μl of M-MLV reverse transcriptase (200 U/μl; Invitrogen, Karlsruhe, Germany), and DEPC-treated H2O was added to a total volume of 30.2 µl and incubated 10 minutes at 25°C followed by incubation at 42°C for 60 minutes in the thermocycler. The cDNA was stored at -20°C until further use.
Real-time quantitative PCR (RT-qPCR)
RT-qPCR was performed in an ABI Prism 7300 sequence detector (Applied Biosystems, Weiterstadt, Germany). In each reaction, 0.75 µl of cDNA was amplified in a 22 µl volume using the RT-qPCR Core Kit for SYBR Green (Eurogentec, Seraing, Belgium). The reactions were run for 40 circles, including 96°C for 15 seconds and 60°C for 1 minute. The following primer sequences were used: Ccl2 fwd CCA GAT GCA GTT AAT GCC CC, rev TCT CCA GCC GAC TCA TTG G; Il1b fwd CCA GAT GAG AGC ATC CAG CTT, rev ACA ATG AGT GAC ACT GCC TTC CT; Gapdh fwd ACA AGA TGG TGA AGG TCG GTG, rev AGA AGG CAG CCC TGG TAA CC. GAPDH was used as a housekeeping gene and all the results were calculated using the ∆∆Ct method. Each sample was assayed in duplicate.
Tissue protein extraction
Renal cortex protein was extracted on ice using a protein lysis buffer containing 10 µl protease and phosphatase inhibitors cocktail (Thermo Fisher Scientific, Meerbusch, Germany). After mixing in a vibrator mill for 2-10 minutes, cells were additionally sonicated three times for 10 seconds. The supernatant was collected after centrifugation at 10,000 rpm for 10 minutes at 4°C.
Western Blot and SDS PAGE
Ten micrograms of protein were separated under denaturing conditions on a 4-12% Bis-Tris gel and transferred to a nitrocellulose membrane, then blocked for 1 hour at room temperature with 5% non-fat milk powder in tris-buffered saline and 1% Tween 20 (TTBS). Blots were incubated with the primary antibody overnight at 4°C, washed, and incubated with HRP-labeled secondary antibody for 30 minutes. Bands were detected using enhanced chemiluminescence (ECL) substrate (Thermo Fisher Scientific, Meerbusch, Germany) and visualized using a standard AGFA imaging system (AGFA, Mortsel, Belgium). After chemiluminescent detection, blots were washed and stripped in stripping buffer for 30 minutes at 56°C and reused Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal reference protein. All antibodies used are listed in Table 1. The intensity of the bands was quantified using ImageJ software.
Electron microscopy
Samples prepared for transmission electron microscopy were fixed in a solution containing glutaraldehyde, paraformaldehyde, and sodium-cacodylate. Samples were post-fixed in 1% OsO4 (Carl Roth, Karlsruhe, Germany) and dehydrated by ascending ethanol series (30, 50, 70, 90, and 100%). Dehydrated specimens were incubated in propylene oxide (Serva, Heidelberg, Germany) and embedded in Epon resin (Serva, Heidelberg, Germany). Ultrathin sections were contrast-enhanced by staining with 0.5% uranyl acetate and 1% lead citrate (both from EMS, Munich, Germany) and viewed with a transmission electron microscope (Zeiss Leo906; Carl Zeiss, Oberkochem, Germany). Six to twelve glomeruli per animal were viewed and assessed regarding podocyte stress and thickness of the GBM.
Statistical analyses
p-values < 0.05 were considered as statistically significant. Values are presented as means and standard deviations. Values were analyzed using the Mann-Whitney U test or Kruskal‑Wallis test with post hoc Dunn’s multiple comparisons after exclusion of significant outliers (Grubbs test). The interaction between body weight and serum creatinine was investigated by Spearman correlation. Statistical analyses were performed using GraphPad Prism Version 7 (GraphPad Software, San Diego, CA).
Results
All animals that survived till the end of the study (14 months) were used for the analyses and no animals or data were excluded. The only exception was the ultrastructural analyses using electron microscopy, which, due to its laborious nature was performed in two animals per group (one male and one female). Statistical comparisons were performed for all animals together as well as separately for females and males.
HFHCD-induced glucose intolerance and visceral obesity without a decline in kidney function
All animals except for females in the HFHCD+liraglutide groups showed a significant increase in waist circumference during the five month intervention phase (Fig 1B+B’).
The comparison of the four treatment groups at the time of the end of the study showed that the HFHCD group had a trend towards higher body weight than the control group even though in the control group animals consumed higher amounts of the normal caloric diet. Weight gain was less pronounced in both the HFHCD+liraglutide and HFHCD+metformin groups (Table 2). Overall, changes in body weight remained non-significant in all three HFHCD groups compared to the control group (Table 2). Taken together, the HFHCD animals did not develop severe obesity but signs of visceral obesity, as indicated by an increase of waist circumference, were observed.
Serum cholesterol values were comparable in all groups. While most groups did not show a statistically different concentration of serum glucose, animals, and particularly males treated with HFHCD+liraglutide, exhibited significantly higher serum glucose values compared to animals in the control and HFHCD group (Table 2). In general, animals on HFHCD developed glucose intolerance as described elsewhere (Heffer et al., manuscript in preparation).
Blood urea nitrogen (BUN) levels were significantly lower in males of the HFHCD- and HFHCD+metformin groups compared to the control group while there were no differences in female animals (Fig. 1C+C’, Table 3). Serum creatinine values and albuminuria did not significantly differ between the groups (Table 3). The kidney to body weight ratio was comparable in all intervention groups. Gender-specific analyses revealed that females in the HFHCD+liraglutide group had a significantly higher body weight/kidney weight ratio than the other three female intervention groups (Table 3).
There was a moderate negative correlation (Spearman ρ = -0.5) between the body weight and the serum creatinine levels (Fig. 1D). No significant differences in overall survival were found (p = 0.36; Fig. 1E).
Taken together, six months of HFHCD did not lead to any significant changes in kidney function. Since the functional kidney parameters are not sensitive for early kidney injury, detailed histomorphological and molecular analyses of the kidneys were performed.
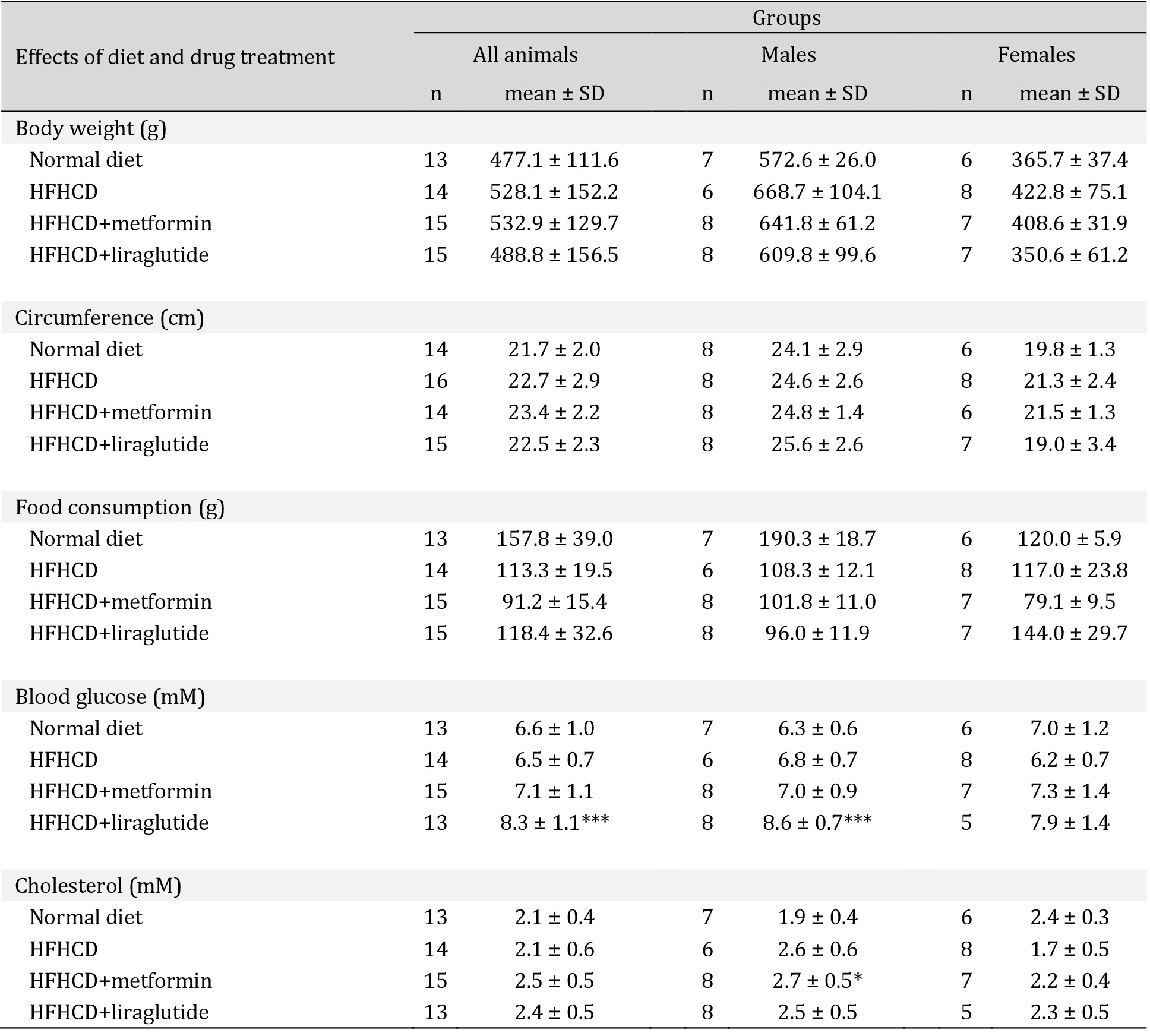
Basic characteristics and diet effects in all treatment groups after 19 weeks (at the end of the experiment or the last sampling before sacrifice)- Effects of diet and drug treatment. BUN = blood urea nitrogen, HFHCD = high-fat-high-carbohydrate diet, n = number, SD = standard deviation * p<0.05, ** p<0.01, *** p<0.001; **** p<0.0001 versus normal diet
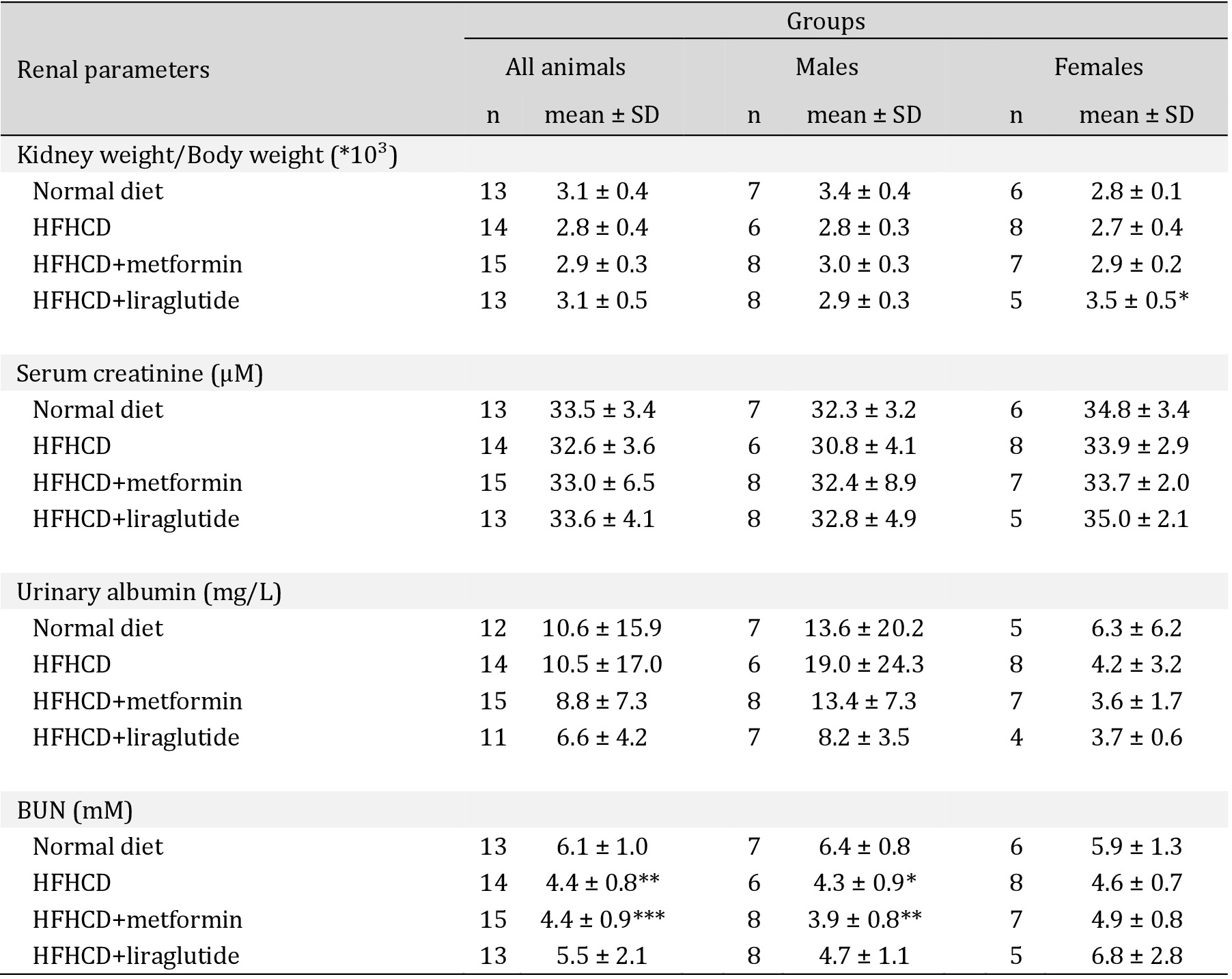
Basic characteristics and diet effects in all treatment groups after 19 weeks (at the end of the experiment or the last sampling before sacrifice)- Renal parameters. BUN = blood urea nitrogen, HFHCD = high-fat-high-carbohydrate diet, n = number, SD = standard deviation * p<0.05, ** p<0.01, *** p<0.001; **** p<0.0001 versus normal diet
Six months of HFHCD did not lead to glomerular hypertrophy or glomerular scar formation
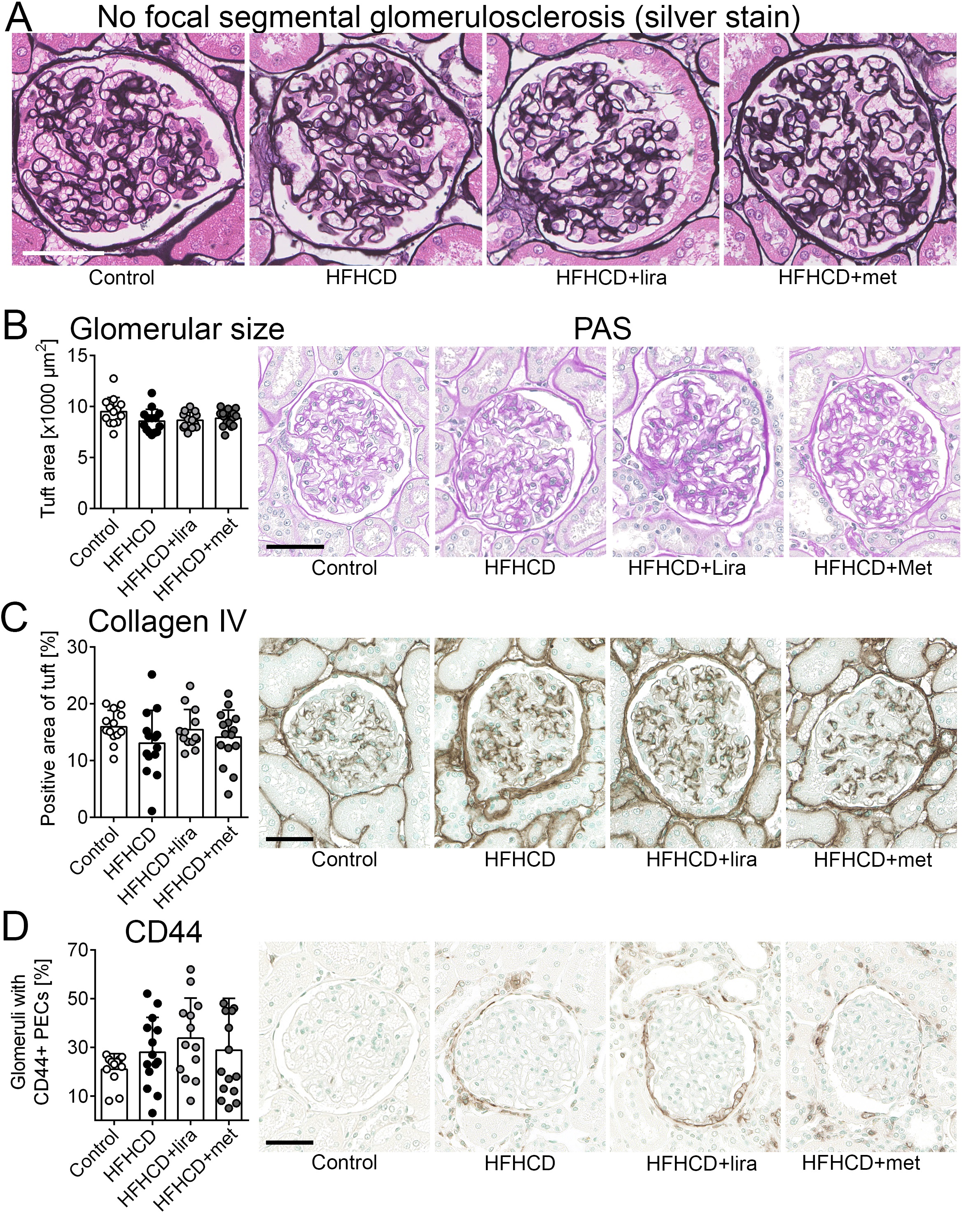
First, structural glomerular changes within the control and the three intervention groups were examined using silver stain. In all animals, there were neither signs of basement membrane thickening nor signs of adhesions of GBMs to Bowman’s capsule, as the initial stage of FSGS development (Fig. 2A). Additionally, no FSGS was found.
Glomerular hypertrophy was analyzed by quantification of glomerular tuft area in PAS-stained slides. There was no significant difference between any of the groups (Fig. 2B) and no gender-related differences were observed (Supplementary Table 1 – for all supplementary material see www.cellphysiolbiochem.com).
Next to histopathological assessment, glomerulosclerosis was analyzed using collagen IV staining which also revealed no significant differences between the groups (Fig. 2C), also if analyzed separately in males and females (Supplementary Table 1).
CD44 is a marker of activated parietal epithelial glomerular cells (PECs) and its de novo expression in PECs is considered to be a precursor lesion in FSGS development [32, 33]. Glomeruli with positively stained PECs were counted and no significant differences between the groups were detected. However, the control group had the lowest number of CD44 positive glomeruli (Fig. 2D). In males, the number of CD44 positive glomeruli was significantly higher in the HFHCD group than in the control group (p = 0.03). In females, there were no significant changes between all groups (Supplementary Table 1).
Taken together, no major signs of glomerular hypertrophy or FSGS were observed across the intervention groups. Only male HFHCD animals showed significantly more activated PECs as FSGS precursor lesions compared to males of the control group.
Six months of HFHCD led to a significant glomerular downregulation of the intermediate filament nestin
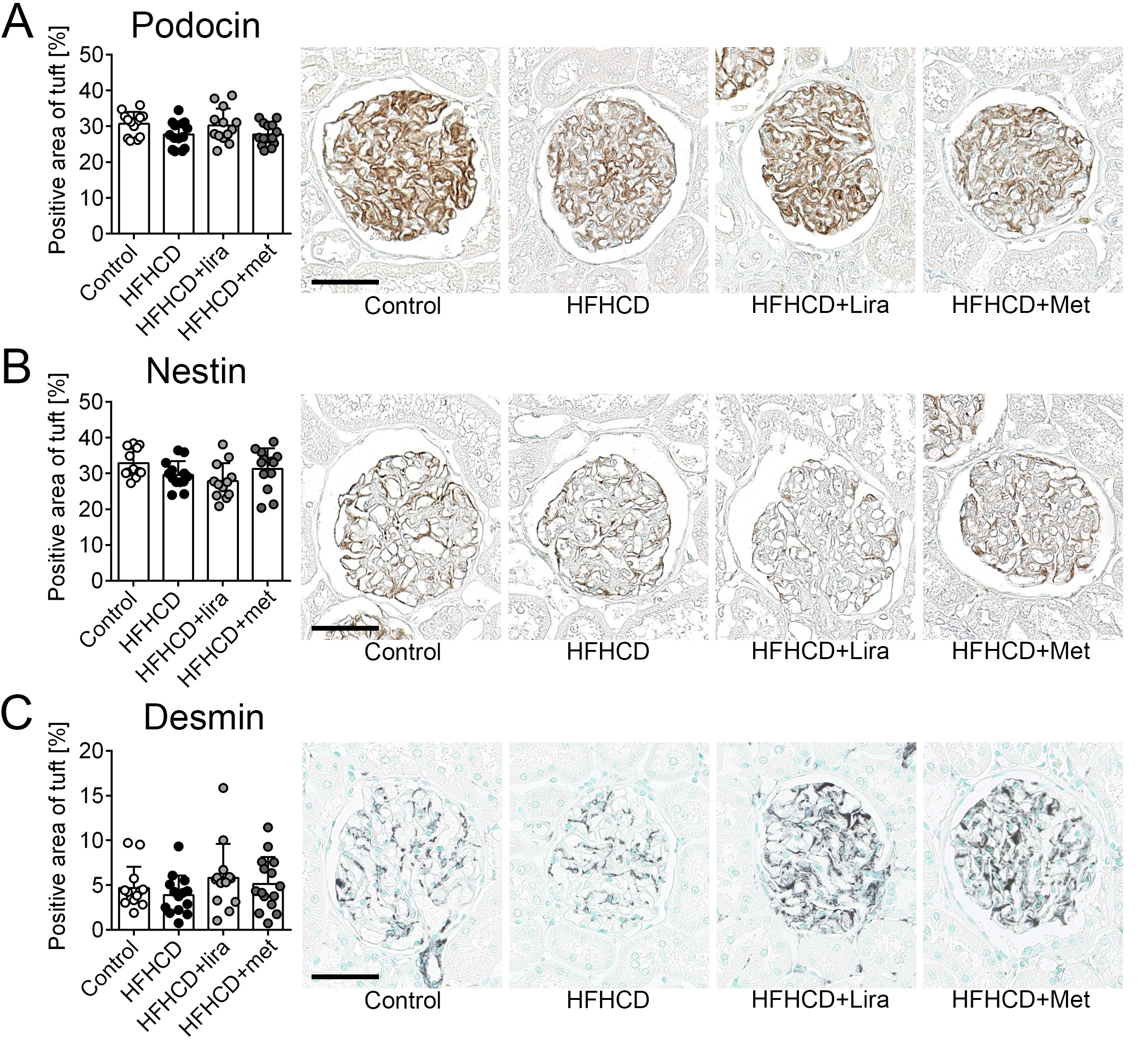
The glomerular expression of podocin, a specific slit diaphragm protein that is often reduced during podocyte damage, was similar in all groups (Fig. 3A), and no significant gender-specific differences were observed (Supplementary Table 1). Additionally, the podocyte phenotype was investigated by stainings of the intermediate filaments nestin and desmin. Nestin was downregulated in podocytes of patients with different glomerular
diseases [34]. The nestin positive area was significantly lower in the HFHCD and HFHCD+liraglutide groups compared to the control group. The HFHCD+liraglutide group also exhibited less nestin expression compared to the HFHCD+metformin group (Fig. 3B).
In a gender-stratified analysis, females of the control group had a significantly higher nestin-positive area than females of the HFHCD and HFHCD+liraglutide groups. The nestin expression in females of the HFHCD+metformin group did not differ from the control group (Supplementary Table 1). In males, no significant intergroup changes could be observed (Supplementary Table 1).
Increased desmin expression is an established marker for podocyte injury [35, 36]. The glomerular positively stained area for desmin was similar among the treatment groups and no gender-specific differences were observed (Fig. 3C, Supplementary Table 1).
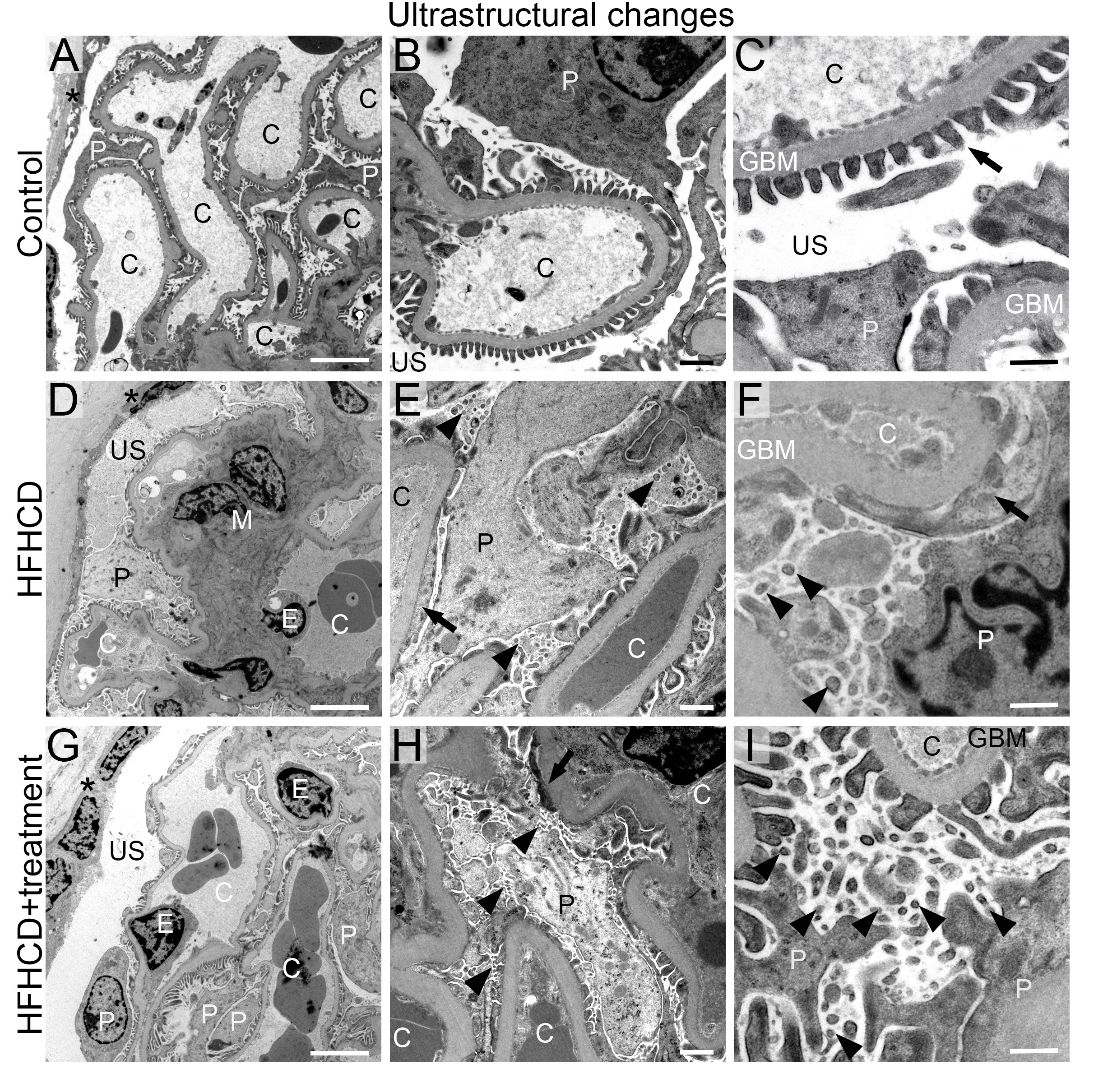
Since no overt pathological changes were observed, but the molecular analyses suggested possible podocyte injury, transmission electron microscopy was performed in two representative animals per control and intervention group (one male, one female) to investigate effects on the podocyte ultrastructure (Fig. 4 (males), Supplementary Fig. 1 (females)). The control group showed normal glomerular structure with open capillaries, fenestrated endothelium, regular mesangium, and regular podocyte foot processes (Fig. 4A-C, Supplementary Fig. 1A-C). In contrast, animals of the HFHCD, the HFHCD+liraglutide, and the HFHCD+metformin groups exhibited mild podocyte stress as shown by a segmental podocyte foot process effacement, with approx. 10% of the foot processes being affected, and a prevalent formation of microvilli in almost all podocytes (Fig. 4D-I, Supplementary Fig. 1D-I). The glomerular tufts in the HFHCD group were denser because of mild mesangial hypercellularity and mesangial matrix expansion compared to the HFHCD+liraglutide and HFHCD+metformin groups (Fig. 4D). GBM sizes were similar in the control and the intervention groups.
Taken together, HFHCD-induced podocyte damage was observed as shown by podocyte nestin expression and ultrastructural morphology. Treatment with liraglutide did not significantly improve the podocyte expression of the intermediate filament nestin, while the nestin expression in the HFHCD+metformin group was unchanged compared to the control group.
Tubulointerstitial damage and signs of inflammation
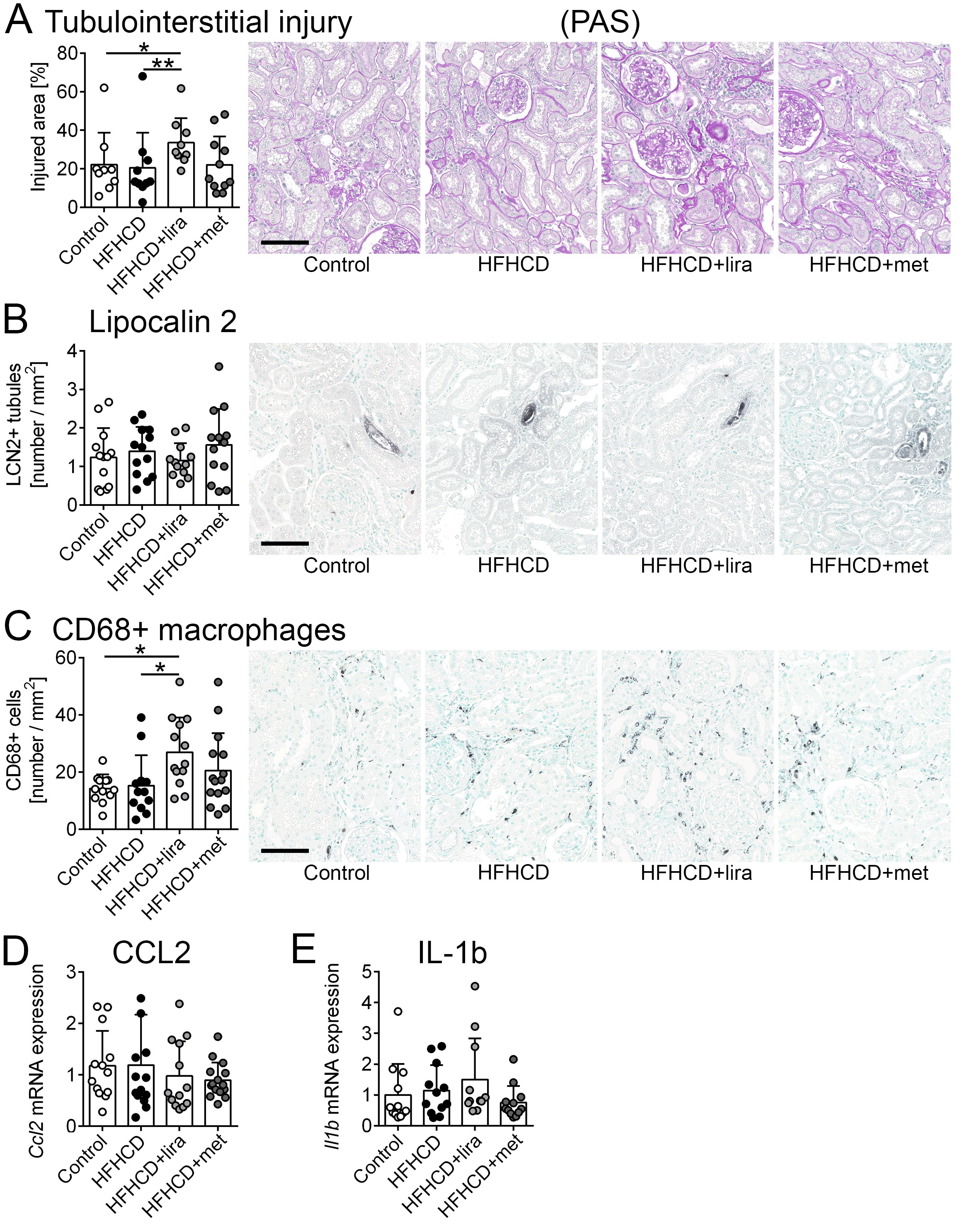
Apart from age-associated tubulointerstitial changes, there were only minor differences in tubular injury between the groups (Fig. 5). The HFHCD+liraglutide group exhibited significantly higher tubulointerstitial damage compared to the HFHCD group and the control group when analyzed as a percentage of the affected cortical area (PAS staining, Fig. 5A). A separate analysis of males and females did not reveal significant changes between the four treatment groups (Supplementary Table 2).
The tubular injury marker lipocalin-2 was not regulated (Fig. 5B) and no gender-related differences were found (Supplementary Table 2). Additionally, CD44 and CD68 were used as markers for immune cells in the tubulointerstitium. Both the percentage of the CD44 positive stained area (Supplementary Fig. 2) and the amount of CD68 positive cells (Fig. 5C) were similar between the control and the HFHCD groups, but significantly higher in the HFHCD+liraglutide group compared to the control and (for CD68) also in the HFHCD group.
Additionally, renal inflammation was analyzed by mRNA expression of CCL2 and IL-1b. Both cytokines did not show any significant differences between the groups and no gender-specific changes were observed (Fig. 5D-E; Supplementary Table 2).
Taken together, we found slightly higher tubulointerstitial damage and immune cell infiltration in the HFHCD+liraglutide group compared to the control and the HFHCD groups.
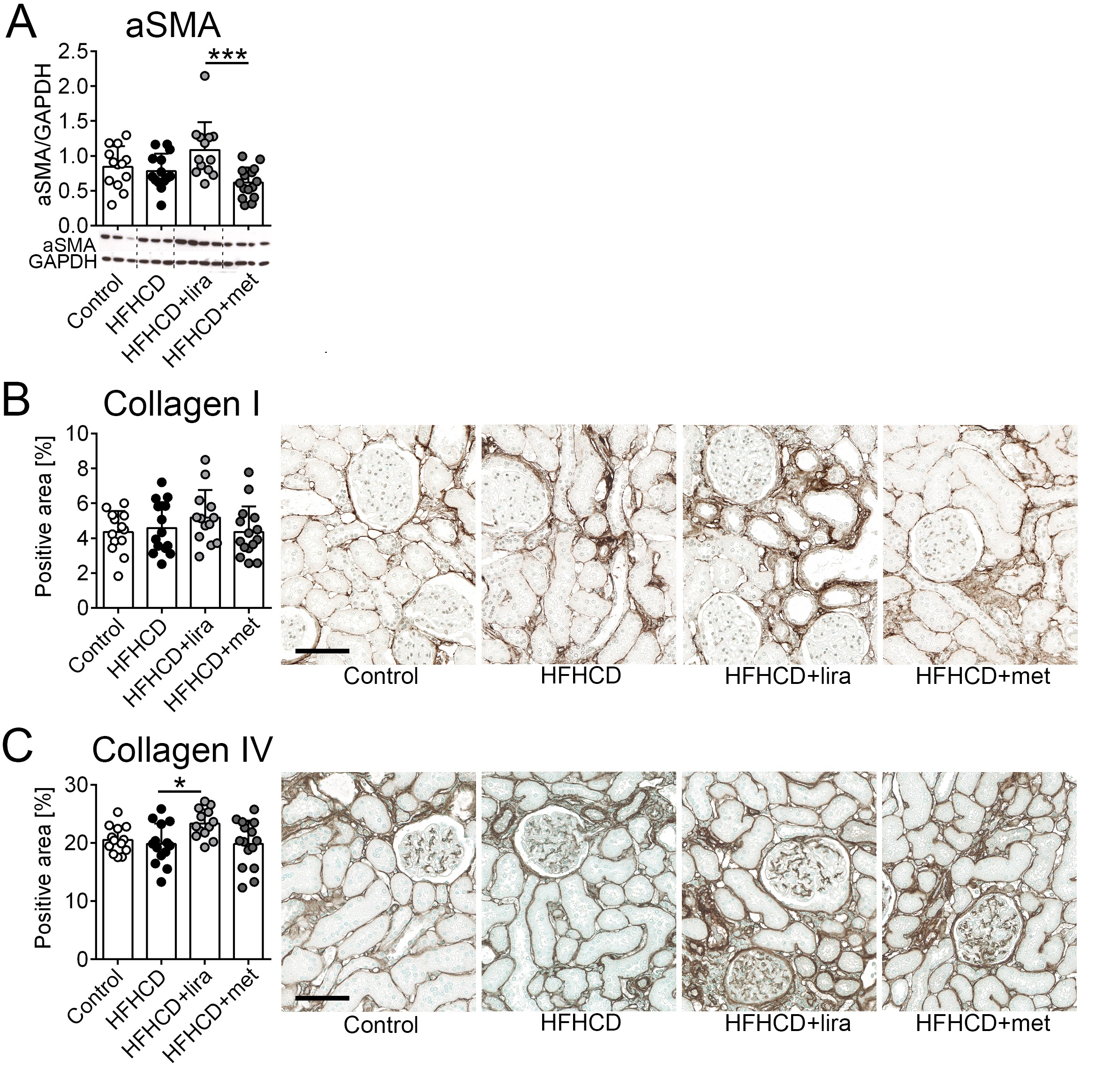
Tubulointerstitial fibrosis
Alpha smooth muscle actin (aSMA), which is expressed by myofibroblasts and used as a marker of fibrogenesis, did not show any difference between the control and the three intervention groups. The gender‑stratified analysis revealed that only in male animals, the aSMA values of the HFHCD+liraglutide group were significantly higher compared to the HFHCD+metformin group but not to the control or the HFHCD groups (Fig. 6A, Supplementary Table 2).
The collagen I positive interstitial area was similar between the groups and no gender-specific differences were observed (Fig. 6B, Supplementary Table 2).
Collagen type IV expression was not elevated in the HFHCD group compared to the control group. Still, collagen type IV was elevated in the HFHCD+liraglutide treated group compared to those of the HFHCD group (Fig. 6C). The gender-stratified analysis showed that the higher collagen IV expression in kidneys of the HFHCD+liraglutide group was driven by male animals (Supplementary Table 2).
Taken together, HFHCD did not promote fibrogenesis, however, additional treatment with liraglutide led to a slight, but significantly increased interstitial microinflammation, fibroblast activation, and extracellular matrix accumulation, which was more pronounced in male animals.
Discussion
Here we analyzed a preclinical model that was designed to mimic elderly patients with longstanding obesity and metabolic syndrome. Most of the preclinical models which are currently used to study obesity and metabolic syndrome-related renal effects use young animals with genetic mutations in the leptin signaling pathway. Some of the best-known strains to model obesity in rats are leptin and leptin receptor dysfunctional models, e.g., the obese Zucker rat [37, 38]. This strain was discovered in the early 1960s and has been further developed by crossing with several other strains to create a multitude of models, e.g., the DahlS.Z-Leprfa/Slc rat in 2011 [39]. Commonly, these strains develop obesity and metabolic syndrome early in life. Surprisingly, obese Zucker rats do not develop severe kidney damage but rather mild damage despite overt obesity [40]. Kidney damage is also correlated with aging and not solely with obesity [41]. While leptin resistance has occasionally been observed in humans, dietary and environmental factors are crucial for the development of obesity. We here analyzed a non-genetic model of metabolic syndrome using a long-term HFHCD diet in rats. The major advantages of our model are that it does not use genetically modified animals, uses aged rats (9 months), and a long-term dietary intervention (5 months on HFHCD diet), overall, better reflecting the clinical situation.
Despite the long feeding with a HFHCD in aged rats, the animals did not develop severe kidney damage. One reason might be that the animals showed only mild signs of metabolic syndrome, with mild visceral obesity and insulin resistance. Dyslipidemia, severe obesity, and overt diabetes did not occur. Given these findings, we conclude that our model is not suitable to study the effects of overt metabolic syndrome but might render itself suitable to study the mechanisms that contribute to early podocyte injury.
Glomerular hyperfiltration is commonly observed early in patients with morbid obesity and is characterized by glomerular hypertrophy [11, 42]. While we were able to notice a moderate negative correlation (Spearman ρ = -0.5) between the body weight and the serum creatinine levels potentially indicating signs of the beginning of glomerular hyperfiltration, we failed to detect glomerular hypertrophy, thickening of the GBM or FSGS across the treatment groups. On the other hand, nestin was significantly downregulated in animals of the HFHCD group. Recently nestin was described as a predictive factor for the development of FSGS in children [43] and in the absence of nestin also podocyte effacement may appear [44]. Together with the findings of ultrastructural signs of podocyte injury, these results point to early podocyte damage caused by the HFHCD in this model. This suggests, that potential treatments targeting the podocytes, might be important long before there are marked alterations of renal function or structural damage, e.g., FSGS.
In other studies, liraglutide was proven to act in an anti-inflammatory, anti-fibrotic and renoprotective manner [19, 21, 45-47]. Here, we found a significant increase in CD44 and CD68 positive inflammatory cells and a more pronounced tubulointerstitial fibrosis in the HFHCD+liraglutide treated group compared to the HFHCD group. It is tempting to hypothesize that both drugs, but particularly liraglutide, might not be effective as prevention or early intervention in the initial stage of kidney disease, as we have observed in our model. Still, we cannot draw further conclusions from this study, as the animals did not develop a full metabolic syndrome including obesity and hyperglycemia. The observed mild tubulointerstitial changes thus might not have disease-relevant pathological consequences.
A secondary aspect of this study was to evaluate potential gender-related differences and effects following the HFHCD diet and the treatments. It is known that men with type I
diabetes mellitus have a higher risk to develop diabetic nephropathy than women [48]. There are also gender-specific differences in the prediction of cardiovascular diseases resulting from obesity and metabolic syndrome [49]. In our gender-specific analyses, we could observe that the first signs of glomerular damage, i.e., increase of CD44 as a marker for PEC activation, were more pronounced in male animals of the HFHCD group. On the other hand, nestin regulation was mainly driven by female animals of the cohort. This could point to putative gender differences concerning the susceptibility to nutrient-initiated renal damage which is in line with another study which investigated the effects of an early in life exposure to a high-fat diet in male versus female Sprague Dawley rats [50].
Our study has several limitations. Despite the long treatment period, animals did not develop overt obesity and diabetes. Hence, we presume that the duration of HFHCD was probably insufficient to develop clear signs of obesity and metabolic syndrome-related kidney damage. One former study used very large group sizes (380 rats overall) and a very long period of 26 months of the diet, but an increase of nephrosclerosis was only described after 13 months of treatment [51]. Although we had already chosen relatively large group sizes, they may not have been large enough to detect significant changes after 6 months of HFHCD. However, other studies have also shown effects with smaller group sizes and shorter treatment durations, which we could not confirm in our study [52]. Interestingly, even the initial aging process did not lead to increased renal damage under HFHCD. It is possible that an earlier onset of HFHCD could further influence the extent of renal damage or that another rat strain would be more susceptible [41]. Other studies have shown that a long-term high fat diet led to the development of obese but otherwise healthy male and female Sprague-Dawley rats [53] and that additional second-hit factors, e.g., hypertension and streptozotocin injections were required to mimic metabolic syndrome and signs of established diabetic nephropathy [54]. However, these studies analyzed young(er) rats and have not perfomed detailed structural analyses including electron microscopy.
Conclusion
Compared to control animals, we found signs of early and mild podocyte stress in animals fed with HFHCD. Our model could be useful to characterize early signs of obesity and metabolic syndrome-induced podocyte damage.
We thank Simon Wilhelm Otten, Jana Baues, Christina Gianussis and Marie Cherelle Timm for excellent technical assistance.
Author Contributions
The study was designed by B.M.K., P.B., S.G.V., C.S. and R.G. The animal model has been performed by S.G.V., R.G., E.D., A.S-I., A.B.S and K.S. Renal histology, immunohistochemistry, RNA expression and statistical analyses have been performed and evaluated by E.L., B.M.K. and C.S.. E.M.B. performed electron microscopy. C.S. wrote the manuscript. B.M.K. created the figures. B.M.K. and P.B. edited the manuscript. Supervision was given by R.G., S.G.V., J.F., B.M.K. and P.B. Each author contributed important intellectual input during manuscript drafting, accepted personal accountability for the author’s own contributions, and agreed to ensure that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. The final manuscript was approved by all listed authors.
Funding
This study was funded by the German Research Foundation (DFG, Project-IDs 322900939 – SFB TRR 219, 454024652, 432698239 to PB; and 445703531 - CRU 5011 IntraKD to PB and BMK), the Federal Ministry of Education and Research (STOP-FSGS-01GM1901A to PB), and by grants from the Medical Faculty of the RWTH Aachen (START 109/20 to BMK).
This work was also supported by the Association for Regional Cooperation in the Fields of Health, the Science and Technology (RECOOP HST Association) RECOOP Fusion Grant Obesity and Diabetes 2015.
Statement of Ethics
All rat experiments were performed in accordance with the guideline for care and use of laboratory animals and have been approved by local review boards.
The authors declare that no conflict of interests exist.
| 1 Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, et al.: Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014;384:766-781. https://doi.org/10.1016/S0140-6736(14)60460-8 |
||||
| 2 Wahba IM, Mak RH: Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol 2007;2:550-562. https://doi.org/10.2215/CJN.04071206 |
||||
| 3 Bonnet F, Deprele C, Sassolas A, Moulin P, Alamartine E, Berthezene F, Berthoux F: Excessive body weight as a new independent risk factor for clinical and pathological progression in primary IgA nephritis. Am J Kidney Dis 2001;37:720-727. https://doi.org/10.1016/S0272-6386(01)80120-7 |
||||
| 4 Chintam K, Chang AR: Strategies to Treat Obesity in Patients With CKD. Am J Kidney Dis 2020;77:427-439. https://doi.org/10.1053/j.ajkd.2020.08.016 |
||||
| 5 Kambham N, Markowitz GS, Valeri AM, Lin J, D'Agati VD: Obesity-related glomerulopathy: an emerging epidemic. Kidney Int 2001;59:1498-1509. https://doi.org/10.1046/j.1523-1755.2001.0590041498.x |
||||
| 6 Kwakernaak AJ, Zelle DM, Bakker SJ, Navis G: Central body fat distribution associates with unfavorable renal hemodynamics independent of body mass index. J Am Soc Nephrol 2013;24:987-994. https://doi.org/10.1681/ASN.2012050460 |
||||
| 7 Okabayashi Y, Tsuboi N, Sasaki T, Haruhara K, Kanzaki G, Koike K, Shimizu A, D'Agati VD, Yokoo T: Single-Nephron GFR in Patients With Obesity-Related Glomerulopathy. Kidney Int Rep 2020;5:1218-1227. https://doi.org/10.1016/j.ekir.2020.05.013 |
||||
| 8 Denic A, Glassock RJ: Obesity-Related Glomerulopathy and Single-Nephron GFR. Kidney Int Rep 2020;5:1126-1128. https://doi.org/10.1016/j.ekir.2020.05.017 |
||||
| 9 Kurella M, Lo JC, Chertow GM: Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol 2005;16:2134-2140. https://doi.org/10.1681/ASN.2005010106 |
||||
| 10 Oterdoom LH, de Vries AP, Gansevoort RT, de Jong PE, Gans RO, Bakker SJ: Fasting insulin modifies the relation between age and renal function. Nephrol Dial Transplant 2007;22:1587-1592. https://doi.org/10.1093/ndt/gfm037 |
||||
| 11 D'Agati VD, Chagnac A, de Vries AP, Levi M, Porrini E, Herman-Edelstein M, Praga M: Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol 2016;12:453-471. https://doi.org/10.1038/nrneph.2016.75 |
||||
| 12 Tsuboi N, Okabayashi Y, Shimizu A, Yokoo T: The Renal Pathology of Obesity. Kidney Int Rep 2017;2:251-260. https://doi.org/10.1016/j.ekir.2017.01.007 |
||||
| 13 Tack CJ, Jacob S, Desouza C, Bain SC, Buse JB, Nauck MA, Petrie JR, Poulter NR, Pratley RE, Stegmann H, Bosch-Traberg H, Startseva E, Zinman B: Long-term efficacy and safety of combined insulin and glucagon-like peptide-1 therapy: Evidence from the LEADER trial. Diabetes Obes Metab 2019;21:2450-2458. https://doi.org/10.1111/dom.13826 |
||||
| 14 Thethi TK, Pratley R, Meier JJ: Efficacy, safety and cardiovascular outcomes of once-daily oral semaglutide in patients with type 2 diabetes: The PIONEER programme. Diabetes Obes Metab 2020;22:1263-1277. https://doi.org/10.1111/dom.14054 |
||||
| 15 le Roux CW, Astrup A, Fujioka K, Greenway F, Lau DCW, Van Gaal L, Ortiz RV, Wilding JPH, Skjøth TV, Manning LS, Pi-Sunyer X: 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double-blind trial. Lancet 2017;389:1399-1409. | ||||
| 16 Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, Lau DC, le Roux CW, Violante Ortiz R, Jensen CB, Wilding JP: A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N Engl J Med 2015;373:11-22. https://doi.org/10.1056/NEJMoa1411892 |
||||
| 17 Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML, Woo V, Hansen O, Holst AG, Pettersson J, Vilsbøll T: Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med 2016;375:1834-1844. https://doi.org/10.1056/NEJMoa1607141 |
||||
| 18 Mann JFE, Ørsted DD, Brown-Frandsen K, Marso SP, Poulter NR, Rasmussen S, Tornøe K, Zinman B, Buse JB: Liraglutide and Renal Outcomes in Type 2 Diabetes. N Engl J Med 2017;377:839-848. https://doi.org/10.1056/NEJMoa1616011 |
||||
| 19 Moellmann J, Klinkhammer BM, Onstein J, Stöhr R, Jankowski V, Jankowski J, Lebherz C, Tacke F, Marx N, Boor P, Lehrke M: Glucagon-Like Peptide 1 and Its Cleavage Products Are Renoprotective in Murine Diabetic Nephropathy. Diabetes 2018;67:2410-2419. https://doi.org/10.2337/db17-1212 |
||||
| 20 Sukumaran V, Tsuchimochi H, Sonobe T, Shirai M, Pearson JT: Liraglutide Improves Renal Endothelial Function in Obese Zucker Rats on a High-Salt Diet. J Pharmacol Exp Ther 2019;369:375-388. https://doi.org/10.1124/jpet.118.254821 |
||||
| 21 Kodera R, Shikata K, Kataoka HU, Takatsuka T, Miyamoto S, Sasaki M, Kajitani N, Nishishita S, Sarai K, Hirota D, Sato C, Ogawa D, Makino H: Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia 2011;54:965-978. https://doi.org/10.1007/s00125-010-2028-x |
||||
| 22 He J, Yuan G, Cheng F, Zhang J, Guo X: Mast Cell and M1 Macrophage Infiltration and Local Pro-Inflammatory Factors Were Attenuated with Incretin-Based Therapies in Obesity-Related Glomerulopathy. Metab Syndr Relat Disord 2017;15:344-353. https://doi.org/10.1089/met.2017.0057 |
||||
| 23 Liang R, Wang M, Fu C, Liang H, Deng H, Tan Y, Xu F, Cai M: Liraglutide protects against high-fat diet-induced kidney injury by ameliorating apoptosis. Endocr Connect 2020;9:946-954. https://doi.org/10.1530/EC-20-0294 |
||||
| 24 Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352:854-865. https://doi.org/10.1016/S0140-6736(98)07037-8 |
||||
| 25 Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM: Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393-403. https://doi.org/10.1056/NEJMoa012512 |
||||
| 26 Seifarth C, Schehler B, Schneider HJ: Effectiveness of metformin on weight loss in non-diabetic individuals with obesity. Exp Clin Endocrinol Diabetes 2013;121:27-31. https://doi.org/10.1055/s-0032-1327734 |
||||
| 27 Apolzan JW, Venditti EM, Edelstein SL, Knowler WC, Dabelea D, Boyko EJ, Pi-Sunyer X, Kalyani RR, Franks PW, Srikanthan P, Gadde KM: Long-Term Weight Loss With Metformin or Lifestyle Intervention in the Diabetes Prevention Program Outcomes Study. Ann Intern Med 2019;170:682-690. https://doi.org/10.7326/M18-1605 |
||||
| 28 De Broe ME, Kajbaf F, Lalau JD: Renoprotective Effects of Metformin. Nephron 2018;138:261-274. https://doi.org/10.1159/000481951 |
||||
| 29 Langer S, Kreutz R, Eisenreich A: Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. J Nephrol 2016;29:765-773. https://doi.org/10.1007/s40620-015-0258-1 |
||||
| 30 Zhai L, Gu J, Yang D, Wang W, Ye S: Metformin Ameliorates Podocyte Damage by Restoring Renal Tissue Podocalyxin Expression in Type 2 Diabetic Rats. J Diabetes Res 2015;2015:231825. https://doi.org/10.1155/2015/231825 |
||||
| 31 Cavaglieri RC, Day RT, Feliers D, Abboud HE: Metformin prevents renal interstitial fibrosis in mice with unilateral ureteral obstruction. Mol Cell Endocrinol 2015;412:116-122. https://doi.org/10.1016/j.mce.2015.06.006 |
||||
| 32 Smeets B, Kuppe C, Sicking EM, Fuss A, Jirak P, van Kuppevelt TH, Endlich K, Wetzels JF, Gröne HJ, Floege J, Moeller MJ: Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol 2011;22:1262-1274. https://doi.org/10.1681/ASN.2010090970 |
||||
| 33 Smeets B, Moeller MJ: Parietal epithelial cells and podocytes in glomerular diseases. Semin Nephrol 2012;32:357-367. https://doi.org/10.1016/j.semnephrol.2012.06.007 |
||||
| 34 Su W, Chen J, Yang H, You L, Xu L, Wang X, Li R, Gao L, Gu Y, Lin S, Xu H, Breyer MD, Hao CM: Expression of nestin in the podocytes of normal and diseased human kidneys. Am J Physiol Regul Integr Comp Physiol 2007;292:R1761-1767. https://doi.org/10.1152/ajpregu.00319.2006 |
||||
| 35 Floege J, Hackmann B, Kliem V, Kriz W, Alpers CE, Johnson RJ, Kühn KW, Koch KM, Brunkhorst R: Age-related glomerulosclerosis and interstitial fibrosis in Milan normotensive rats: a podocyte disease. Kidney Int 1997;51:230-243. https://doi.org/10.1038/ki.1997.28 |
||||
| 36 Kakimoto T, Okada K, Fujitaka K, Nishio M, Kato T, Fukunari A, Utsumi H: Quantitative analysis of markers of podocyte injury in the rat puromycin aminonucleoside nephropathy model. Exp Toxicol Pathol 2015;67:171-177. https://doi.org/10.1016/j.etp.2014.11.007 |
||||
| 37 Zucker TF, Zucker LM: Fat accretion and growth in the rat. J Nutr 1963;80:6-19. https://doi.org/10.1063/1.3050872 |
||||
| 38 White DW, Wang DW, Chua SC, Jr., Morgenstern JP, Leibel RL, Baumann H, Tartaglia LA: Constitutive and impaired signaling of leptin receptors containing the Gln --> Pro extracellular domain fatty mutation. Proc Natl Acad Sci U S A 1997;94:10657-10662. https://doi.org/10.1073/pnas.94.20.10657 |
||||
| 39 Hattori T, Murase T, Ohtake M, Inoue T, Tsukamoto H, Takatsu M, Kato Y, Hashimoto K, Murohara T, Nagata K: Characterization of a new animal model of metabolic syndrome: the DahlS.Z-Lepr(fa)/Lepr(fa) rat. Nutr Diabetes 2011;1:e1. https://doi.org/10.1038/nutd.2010.1 |
||||
| 40 Coimbra TM, Janssen U, Gröne HJ, Ostendorf T, Kunter U, Schmidt H, Brabant G, Floege J: Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int 2000;57:167-182. https://doi.org/10.1046/j.1523-1755.2000.00836.x |
||||
| 41 McPherson KC, Shields CA, Poudel B, Fizer B, Pennington A, Szabo-Johnson A, Thompson WL, Cornelius DC, Williams JM: Impact of obesity as an independent risk factor for the development of renal injury: implications from rat models of obesity. Am J Physiol Renal Physiol 2019;316:F316-f327. https://doi.org/10.1152/ajprenal.00162.2018 |
||||
| 42 Palatini P: Glomerular hyperfiltration: a marker of early renal damage in pre-diabetes and pre-hypertension. Nephrol Dial Transplant 2012;27:1708-1714. https://doi.org/10.1093/ndt/gfs037 |
||||
| 43 Životić M, Bogdanović R, Peco-Antić A, Paripović D, Stajić N, Vještica J, Ćirović S, Trajković G, Marković-Lipkovski J: Glomerular nestin expression: possible predictor of outcome of focal segmental glomerulosclerosis in children. Pediatr Nephrol 2015;30:79-90. https://doi.org/10.1007/s00467-014-2893-5 |
||||
| 44 Chen J, Boyle S, Zhao M, Su W, Takahashi K, Davis L, Decaestecker M, Takahashi T, Breyer MD, Hao CM: Differential expression of the intermediate filament protein nestin during renal development and its localization in adult podocytes. J Am Soc Nephrol 2006;17:1283-1291. https://doi.org/10.1681/ASN.2005101032 |
||||
| 45 Hendarto H, Inoguchi T, Maeda Y, Ikeda N, Zheng J, Takei R, Yokomizo H, Hirata E, Sonoda N, Takayanagi R: GLP-1 analog liraglutide protects against oxidative stress and albuminuria in streptozotocin-induced diabetic rats via protein kinase A-mediated inhibition of renal NAD(P)H oxidases. Metabolism 2012;61:1422-1434. https://doi.org/10.1016/j.metabol.2012.03.002 |
||||
| 46 Ougaard ME, Sembach FE, Jensen HE, Pyke C, Knudsen LB, Kvist PH: Liraglutide Improves the Kidney Function in a Murine Model of Chronic Kidney Disease. Nephron 2020:1-12. https://doi.org/10.1159/000509418 |
||||
| 47 Li YK, Ma DX, Wang ZM, Hu XF, Li SL, Tian HZ, Wang MJ, Shu YW, Yang J: The glucagon-like peptide-1 (GLP-1) analog liraglutide attenuates renal fibrosis. Pharmacol Res 2018;131:102-111. https://doi.org/10.1016/j.phrs.2018.03.004 |
||||
| 48 Mollsten A, Svensson M, Waernbaum I, Berhan Y, Schon S, Nystrom L, Arnqvist HJ, Dahlquist G, Swedish Childhood Diabetes Study G, Diabetes Incidence Study in S, Swedish Renal R: Cumulative risk, age at onset, and sex-specific differences for developing end-stage renal disease in young patients with type 1 diabetes: a nationwide population-based cohort study. Diabetes 2010;59:1803-1808. https://doi.org/10.2337/db09-1744 |
||||
| 49 Skilton MR, Moulin P, Serusclat A, Nony P, Bonnet F: A comparison of the NCEP-ATPIII, IDF and AHA/NHLBI metabolic syndrome definitions with relation to early carotid atherosclerosis in subjects with hypercholesterolemia or at risk of CVD: evidence for sex-specific differences. Atherosclerosis 2007;190:416-422. https://doi.org/10.1016/j.atherosclerosis.2006.02.019 |
||||
| 50 Moreno JM, Tapia A, Martinez CM, Reverte V, Oltra L, Llinas MT, Salazar FJ: Sex-dependent differences in the adverse renal changes induced by an early in life exposure to a high-fat diet. Am J Physiol Renal Physiol 2019;316:F332-f340. https://doi.org/10.1152/ajprenal.00394.2018 |
||||
| 51 Keenan KP, Coleman JB, McCoy CL, Hoe CM, Soper KA, Laroque P: Chronic nephropathy in ad libitum overfed Sprague-Dawley rats and its early attenuation by increasing degrees of dietary (caloric) restriction to control growth. Toxicol Pathol 2000;28:788-798. https://doi.org/10.1177/019262330002800604 |
||||
| 52 Pan QR, Ren YL, Zhu JJ, Hu YJ, Zheng JS, Fan H, Xu Y, Wang G, Liu WX: Resveratrol increases nephrin and podocin expression and alleviates renal damage in rats fed a high-fat diet. Nutrients 2014;6:2619-2631. https://doi.org/10.3390/nu6072619 |
||||
| 53 Rojas JM, Bolze F, Thorup I, Nowak J, Dalsgaard CM, Skydsgaard M, Berthelsen LO, Keane KA, Søeborg H, Sjögren I, Jensen JT, Fels JJ, Offenberg HK, Andersen LW, Dalgaard M: The Effect of Diet-induced Obesity on Toxicological Parameters in the Polygenic Sprague-Dawley Rat Model. Toxicol Pathol 2018;46:777-798. https://doi.org/10.1177/0192623318803557 |
||||
| 54 Conway BR, Rennie J, Bailey MA, Dunbar DR, Manning JR, Bellamy CO, Hughes J, Mullins JJ: Hyperglycemia and renin-dependent hypertension synergize to model diabetic nephropathy. J Am Soc Nephrol 2012;23:405-411. https://doi.org/10.1681/ASN.2011060577 |
||||