×
![]()
Corresponding Author: Sun Sik Bae
Department of Pharmacology, Pusan National University School of Medicine, Busandaehak-ro 49, Mulgeum-eup, Yangsan-si, Gyungsangnam-do 50612 (Republic of Korea)
Tel. +82-51-510-8065, E-Mail sunsik@pusan.ac.kr
Regulation of Epithelial-Mesenchymal Transition of A549 Cells by Prostaglandin D2
Farzaneh Vafaeinika
Hye Jin Kuma
Seo Yeon Jina
Do Sik Minb
Sang Heon Songc
Hong Koo Had
Chi Dae Kima
Sun Sik Baea
aGene and Cell Therapy Center for Vessel-Associated Disease, Medical Research Institute, and Department of Pharmacology, Pusan National University School of Medicine, Gyungnam, Republic of Korea, bDepartment of Pharmacy, Yeonsei University, Incheon, Republic of Korea, cDepartment of Urology, Pusan National University Hospital, Busan, Republic of Korea, dDepartment of Internal Medicine, Pusan National University Hospital, Busan, Republic of Korea
Introduction
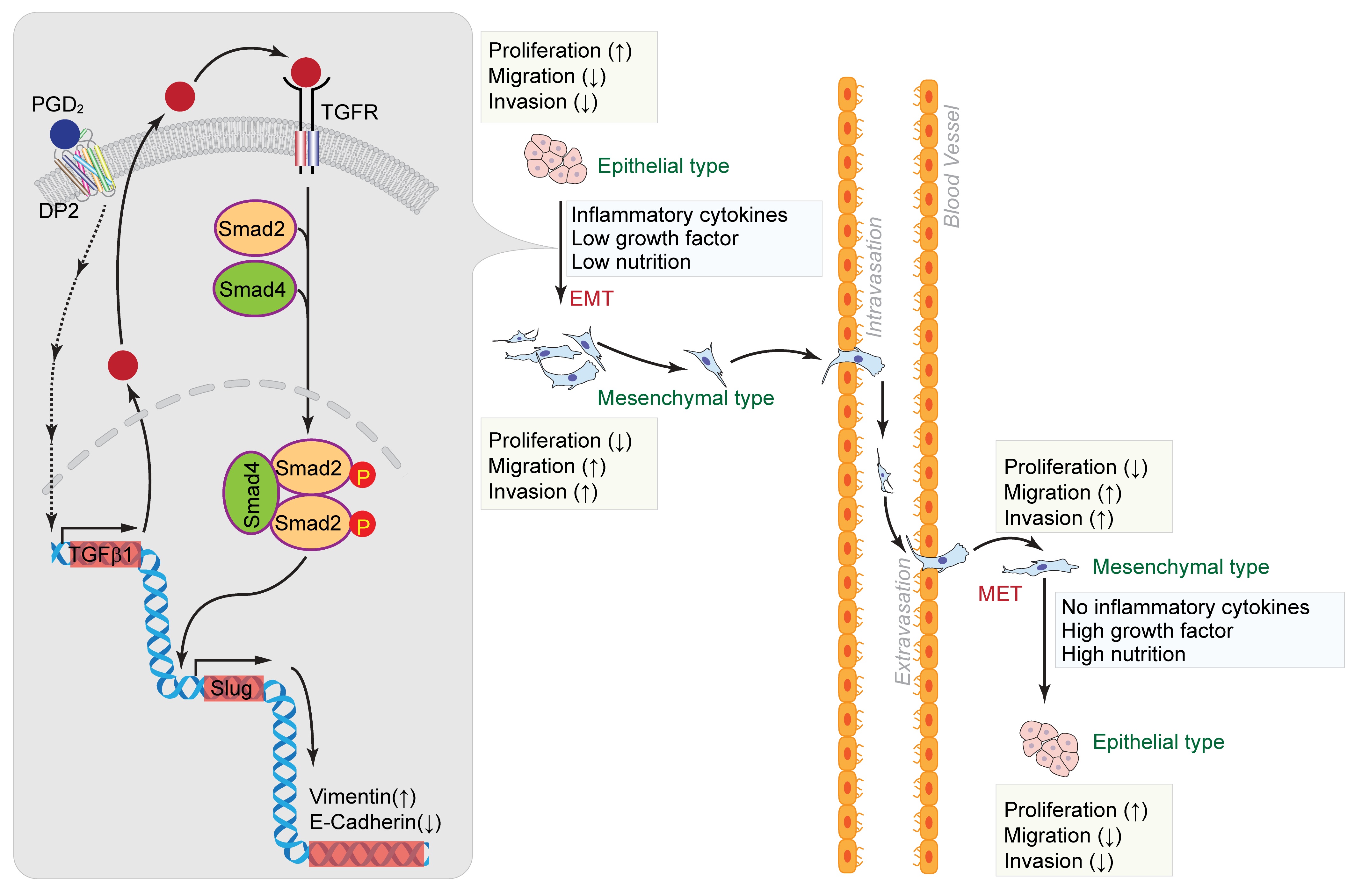
Lung cancer is the most frequent malignancy with the highest mortality rate worldwide [1, 2], and metastasis is the biggest cause of death in patients with lung cancer [3]. Statistically, lung cancer has a higher incidence among men than women [2], and nearly 85% of lung cancer cases are non-small cell lung cancer (NSCLC) [4]. Several current studies have shown that epithelial-mesenchymal transition (EMT) is a significant molecular mechanism that stimulates cancer metastasis [5, 6]. EMT is mainly characterized by the loss of the cell-cell junctions, downregulation of epithelial markers, upregulation of mesenchymal markers, and increased migratory and invasive properties such as the presence of spindle-shaped and motile cells [7, 8]. It also has a critical role in embryonic development, wound healing, and tumor metastasis [9]. During EMT, mesenchymal cells migrate away from the primary tumor and invade blood vessels. Since EMT is a dynamic and flexible process, mesenchymal cells can undergo a reverse process, known as mesenchymal-epithelial transition (MET), reverting to their original epithelial phenotype [8].
In addition, the tumor microenvironment (TME) has a critical role in promoting tumor metastasis and EMT induction. The TME surrounds the primary tumor and is composed of fibroblasts, immune cells, and extracellular matrix (ECM) and is affected by oxygen tension, nutrition, and soluble factors [10]. Within the TME, there are one of the most dynamic cell types or activated fibroblasts called cancer-associated fibroblasts (CAFs), which actively participate in cancer progression. CAFs also possess the ability to regulate EMT in cancer cells [10, 11]. Several factors in the tumor microenvironment directly induce the occurrence of EMT. Particularly, inflammatory cytokines, including transforming growth factor beta 1 (TGF-β1), tumor necrosis factor-α (TNF-α), and interleukin 6 (IL6), are the main factors promoting EMT and tumor invasion. TGF-β1, as the most potent inducer of EMT, stimulates Slug and Twist1 expression in prostate and non-small cell lung cancer. In addition, other cytokines in the TME such as TNF-α and IL6 promote the expression of transcription factors to regulate EMT and tumor progression [10].
In addition to functional programming of tumor cells, new findings have shown that nutrients, which are essential molecules present in the TME, enable tumor cells to adapt to and cope with stressful TME conditions (e.g., nutritional stress, cytokine delivery, acidic and oxidative) [11]. Tumor cells obtain nutrients such as glucose and lipids via the blood supply from the TME and remove some compounds, including protons and lactate, which are immunosuppressive and create changes in macrophage polarization [12]. Enhanced consumption of glucose in tumor cells outsources and impairs T cell function, leading to cancer progression. The nutrients present in the TME cause cancer cells to acquire a more malignant phenotype. Tumor cells can penetrate the surrounding environment and adjust their metabolism [13].
EMT is regulated by various signaling pathways and molecular mechanisms, including TGF-β, Notch, and the Wnt signaling pathways. TGF-β1, a multifunctional cytokine, can induce EMT via the Smad signaling pathway [5]. TGF‐β initiates signaling by forming heteromeric complexes of type 1 (TGFβR1) and type 2 (TGFβR2) receptors. These complexes lead to the activation of Smad2 and Smad3, which further form a complex with Smad4 and translocate to the nucleus. These activated Smad complexes interact with transcription factors such as Slug, ZEB, and Twist to regulate the expression of target genes [14, 15].
Prostaglandins (PGs) are a group of lipid compounds that are enzymatically produced by arachidonic acid through the cyclooxygenase (COX) pathway by two isoforms, COX-1 and COX-2. The conversion of arachidonic acid to prostaglandin H2 (PGH2) is catalyzed by COX (prostaglandin-endoperoxide synthase) enzyme. The respective PG synthase helps in the final conversion of this unstable endoperoxide into individually stable prostaglandin E2 (PGE2), prostaglandin I2 (PGI2), prostaglandin D2 (PGD2), prostaglandin F2 alpha (PGF2α) or thromboxane A2 (TXA2) [16]. Among the different PGs, PGD2 is one of the important COX metabolites [17] and is produced in many organs, including spleen, central nervous system, intestine, and liver [18]. It has been reported that PGD2 has a role in inflammatory responses and sleep promotions [19, 20]. It is reported that the G protein-coupled receptors, D Prostanoid (DP) and the chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) regulate the biological actions of PGD2 [17].
PGD2 is produced primarily by mast cells and to a lesser extent by some other cells like T-helper (Th2) cells, alveolar macrophages and dendritic cells [21, 22]. PGD2 and its metabolites are reported to have an anti-proliferative role in many cell types [18]. It has also been well described that chemotaxis of eosinophils, basophils, and Th2 lymphocytes results in PGD2-promoted enhanced inflammation [23]. Despite the recent insights into the role of PGD2 in inflammation and tumorigenesis, the specific underlying mechanism involved in PGD2-induced tumor formation and metastasis of lung cancer and its role in EMT induction is largely undescribed. Therefore, given the current literature, we used an A549 cell model to examine changes in PGD2-activated EMT-like processes and their role in the migratory and invasive abilities of A549 cells. Furthermore, we explored crosstalk between PGD2 and the TGF-β1 signaling pathways.
Materials and Methods
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), trypsin-EDTA, and antibiotics were purchased from Hyclone Laboratories Inc. (Logan, UT, USA) and Hank’s balanced salt solution (HBSS; Gibco, Cat No: 14175095). Anti-E-Cadherin was purchased from BD Biosciences (San Joes, CA, USA; Cat No: 610181). Anti-Fibronectin was from Abcam (Cambridge, UK, Cat No: ab2413). Anti-Collagen Type I was from Millipore (Bedford, MA, USA, Cat No: AB765P). Anti-actin antibody was obtained from MP Biomedicals (Aurora, OH, USA, Cat No: 691001). Anti-Vimentin was procured from Abcam (Cat No: ab92547). Slug was obtained from GeneTex (Cat No: GTX121924). DAPI, and either Cy3- or Alexa Fluor 488-conjugated goat secondary antibody were purchased from Molecular Probes, Inc. (Carlsbad, CA, USA). IRDye700- and IRDye800-conjugated rabbit/mouse secondary antibodies were obtained from Li-COR Bioscience (Lincoln, NE, USA). Phospho-Smad2 (Ser465/467) (138D4) antibody was from Cell Signaling Technology (Massachusetts, USA, Cat. No: 3108). PGD2 was purchased from Cayman Chemical (Ann Arbor, MI, USA, Cat No: 12010). DK-PGD2 (12610), BW245C (12050) and BWA868C (12060) were procured from Cayman. TM30089 (Cat No: CT-AT002) was obtained from ChemieTek. Recombinant TGF-β1 was procured from R&D systems (Cat. No: 240-B-002) and SB431542 from Sigma-Aldrich (Cat. No: S4317). FastStart Essential DNA Green Master (SYBR green) was purchased from Roche Diagnostics (Indianapolis, IN, USA). All other reagents were high quality and were purchased from Sigma-Aldrich unless otherwise indicated.
Cell culture
A549 cells (a human lung adenocarcinoma cell line) were purchased from Korean Biotech Corp. (Seoul, Korea) and cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin and maintained at 37 °C in 5% CO2. To induce EMT, A549 cells were cultured under the combination of PGD2 (15 µM), low nutrient (HBSS:DMEM = 70%:30%), low serum (2% FBS) condition for 5 days. To induce MET, A549 cells were cultured at 2% FBS, 30% DMEM, and in the presence of PGD2 (15 µM) for 5 days followed by further culture in normal culture condition for 2 days.
Proliferation assay
A549 cells were plated on 6-well plates at a density of 1 × 104 cells in complete medium. Next day, cells were cultured in a low percentage of FBS (0.5%) and in the presence or absence of PGD2. Cells were harvested at the indicated time points and the total number of living cells was counted by using an automatic cell counter (NanoEnTek, Seoul, Korea). To compare the proliferation rates between epithelial type (A549E) and mesenchymal type (A549M) of A549 cells, we first obtained A549M cells by treating A549E cells with PGD2 (15 µM) under low serum condition (0.5% FBS) for 5 days. 1 × 104 cells of both cell types were plated on 6-well plates and cultured at normal culture condition for the indicated period of times, then measured total number of living cells.
Migration assay (Chemotaxis Assay)
Both A549E and A549M cells were cultured and serum-starved for 12 h before plating on a ChemoTx chamber. Cells were detached with trypsin-EDTA and washed with serum-free DMEM. The bottom side of the ChemoTx membrane (8 μm pore size; Neuro Probe Inc., Gaithersburg, MD, USA) was coated with type I collagen overnight, and 1 × 105 cells were over-laid on the upper chamber of the ChemoTx membrane in 50 μL DMEM with 0.5% FBS, and DMEM complete media (10% FBS) was added to the lower chamber. After 4 or 6 h of incubation at 37°C in 5% CO2, the ChemoTx membrane was fixed with 4% paraformaldehyde, and non-migrated cells on the top side of the membrane were removed by gently wiping with a cotton swab. The membrane was stained with DAPI, and migrated cells were counted under a fluorescence microscope at 10× magnitude (Axiovert 200; Carl Zeiss, Jena, Germany).
Invasion assay
Matrigel invasion assays were performed using Matrigel-coated 24-well Transwell plates (BD Biosciences). DMEM complete media (10% FBS) was added to the lower chambers, and 1 × 105 A549E and A549M cells in DMEM with 0.5% FBS were added to the upper chambers. After 24 h, non-migratory cells on the top surface of the filters were wiped off with cotton balls. The cells on the top surface of the filters were wiped off with cotton balls, and migratory cells attached to the lower side of the inserts were fixed with 4% paraformaldehyde (PFA) and was stained with DAPI. Finally, migrated cells were counted under a fluorescence microscope at 10× magnitude (Axiovert 200; Carl Zeiss, Jena, Germany).
Analysis of mRNA expression
The expression of Slug mRNA was measured by real-time quantitative polymerase chain reaction (qPCR) analysis after isolation of total RNA using TRIZOL reagents as described in the manufacturer’s protocol (Invitrogen, Grand Island, NY, USA). One hundred fifty nanograms of total RNA were reverse transcribed into cDNA using ImProm-II reverse transcription systems (Promega Biotec, Madison, WI, USA); the cDNA was then amplified by PCR using specific primers. qPCR data were analyzed using Roche light cycler 96 software (Roche Diagnostics) and the comparative Ct method (2-∆∆CT). Calibration was based on the expression of GAPDH. The primer sequences are shown in Supplementary Table S1 (for all supplementary material see www.cellphysiolbiochem.com).
Short-hairpin RNA and constructs
To silence shTGF-β1 and shTGFβR1, oligonucleotides with an AgeI site at the 5′-end site and a BamHI site at the 3′ end were designed, and sense and antisense oligonucleotides were synthesized (XENOTECH, Daejeon, Korea). Both complementary oligonucleotides were mixed, heated at 98 °C for 5 min, and cooled to room temperature. The annealed nucleotides were subcloned into the AgeI/ BamHI sites of the pLKO.1 lentiviral vector. The shRNA sequences are shown in Supplementary Table S2.
Lentiviral knockdown
For gene silencing, HEK293-FT packaging cells (Invitrogen) were grown to ~70% confluence in six-well plates. The cells were triple transfected with 4 μg of the pLKO.1 lentiviral construct, 1 μg of Δ8.9, and 1 μg of pVSV-G by using a calcium phosphate method. The medium was replaced with fresh medium at 8 h after transfection. Lentiviral supernatants were harvested 24 h and 48 h after transfection and passed through 0.45-μm filters. Cell-free viral culture supernatants were used to infect A549 cells in the presence of 8 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO, USA). Infected cells were isolated for selection by using 10 μg/mL puromycin for 2 days.
Immunocytochemistry
A549 cells were grown on coverslips in 6-well plates cultured for 5 days in the presence or absence of PGD2 (DMEM media containing 0.5% FBS). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS, and incubated anti-E-cadherin or anti-Vimentin overnight, followed by administration of Cy3 or Alexa Flour 488-conjugated secondary antibodies for 2 h, and nuclei were stained with DAPI for 10-15 min. Images were obtained with a confocal microscope at 20× magnification (OLYMPUS FV-1000, Tokyo, Japan).
Western blot
Cells were lysed in 20 mM Tris-HCl, pH 7.4, 1 mM EGTA/EDTA, 1% Triton X-100, 1 mM Na3VO4, 10% glycerol, 1 μg/mL leupeptin and 1 μg/mL aprotinin. Samples were subjected to 8–15% gradient polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. Membranes were incubated with the indicated primary antibodies (Anti-E-Cadherin, Anti-actin, Anti-Vimentin) and IRDye-conjugated secondary antibodies (IRDye700- and IRDye800-conjugated rabbit/mouse), and protein bands were visualized using an Odyssey Infrared Image Analyzer (Li-COR Bioscience).
Statistical analysis
For analysis of mRNA expression, results are expressed as the mean ± SEM of three independent experiments with three replicates each (n = 3). For analysis of proliferation and migration, results are expressed as the mean ± SEM of three independent experiments. When comparing two groups, an unpaired Student’s t-test was used to assess the difference. P-values less than 0.05 were considered significant and indicated as (*). P-values less than 0.01 were designated with two (**). P-values less than 0.001 were shown with three (***), and P-values less than 0.0001 were shown with four (****) asterisks.
Results
PGD 2 induces EMT in A549 cells
As shown in Fig. 1A, stimulation of epithelial type of A549 cells with PGD2 (15 μM) for 5 days induced morphological changes into mesenchymal type. Expression of epithelial marker protein such as E-cadherin was significantly downregulated by the stimulation of A549 cells with PGD2, whereas expression of mesenchymal marker proteins such as Vimentin, Collagen and Fibronectin were significantly upregulated, as determined by western blot (Fig. 1B and Supplementary Fig. S1). The results were also supported by immunocytochemical analyses (Fig. 1C and 1D). In addition, the expression of Slug, which is a key transcriptional regulator of EMT, was significantly enhanced by the stimulation of A549 cells with PGD2 as determined by western blot analysis and quantitative PCR (Fig. 1E and 1F). To verify which receptor is involved in the PGD2-induced EMT of A549 cells, we performed experiments by using specific DP1/DP2 agonists or antagonists. As shown in Fig. 1G, selective agonist for DP2 receptor (DK-PGD2) significantly enhanced EMT of A549 cells whereas selective agonist for DP1 receptor (BW245C) had no effect. In addition, selective antagonist for DP2 receptor (TM30089) significantly blocked PGD2-induced EMT of A549 cells whereas selective antagonist for DP1 receptor (BWA868C) had little effect (Fig. 1H).
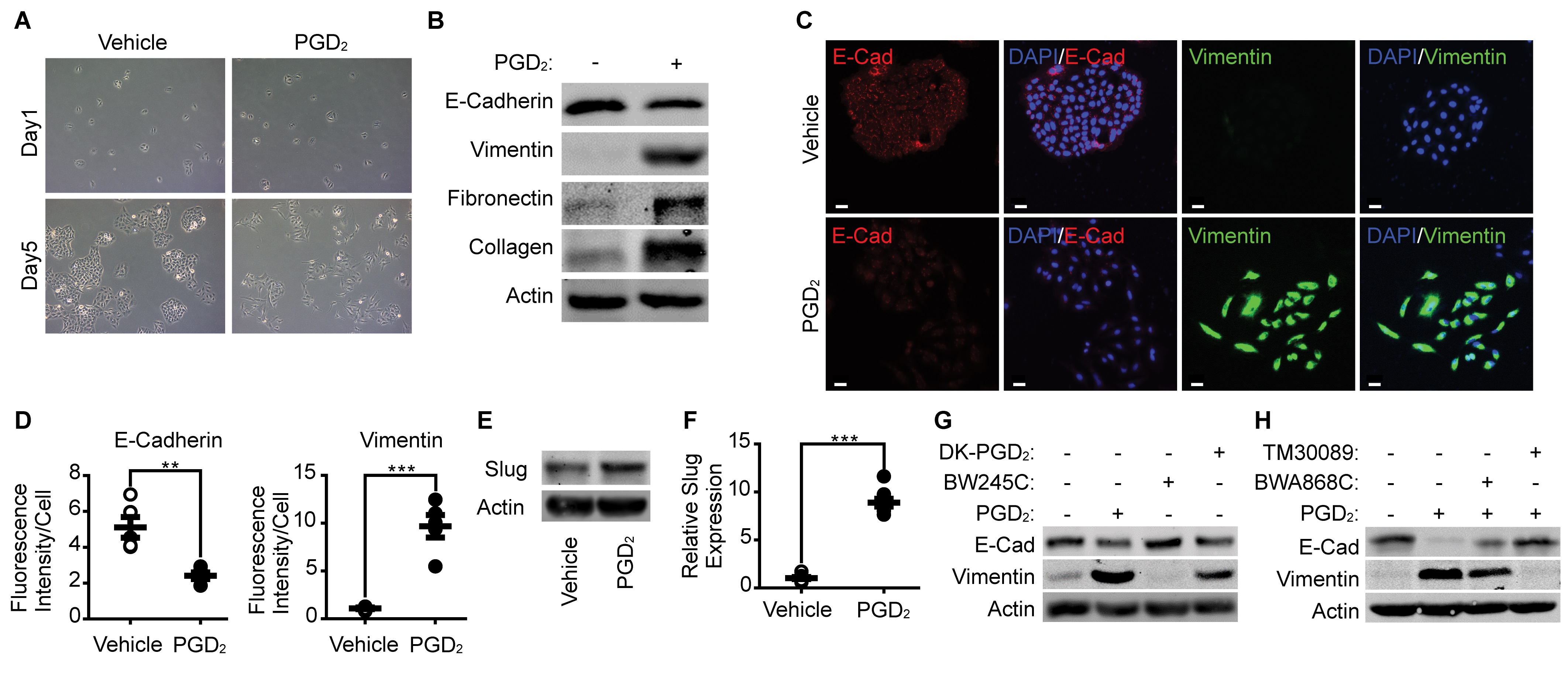
PGD 2 suppresses proliferation and enhances migration & invasion of A549 cells
To evaluate the metastatic properties of A549 cells after stimulation of PGD2, we examined the effect of PGD2 on the proliferation of A549 cells. As shown in Fig. 2A, proliferation of A549 cells was markedly suppressed in the presence of PGD2. To verify whether the suppression of proliferation was caused by EMT during the stimulation of A549 cells with PGD2, we established epithelial type of A549 cells (A549E) and mesenchymal type of A549 cells (A549M) by stimulating A549 cells with PGD2 for 5 days (Fig. 2B). As shown in Fig. 2C, cell proliferation was significantly reduced in A549M cells compared to that in A549E cells. By contrast, the migration and invasion ability was significantly enhanced in A549M cells compared to that in A549E cells (Fig. 2D and 2E).
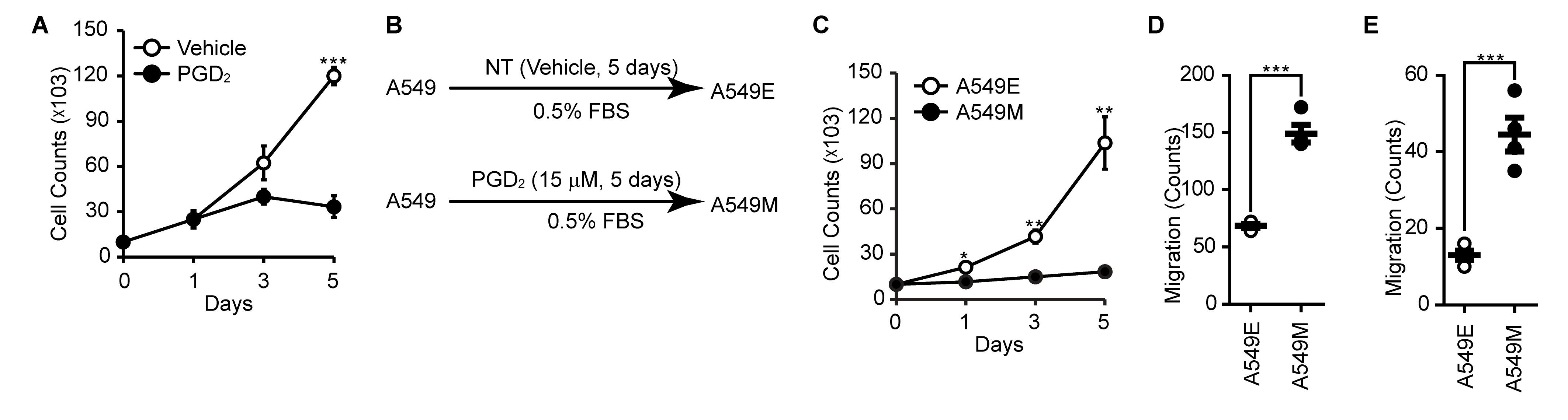
PGD 2 induces EMT through the expression of TGF-β1
Since TGF-β1 is known as major responsive extracellular stimuli, we examined the expression of TGF-β1 in response to PGD2 stimulation. As shown in Fig. 3A and 3B, stimulation of A549 cells with PGD2 significantly upregulated the expression of TGF-β1. In addition, stimulation of A549 cells with PGD2 (Fig. 3C, upper panel) as well as TGF-β1 (Fig. 3C, lower panel) significantly induced the phosphorylation of Smad2 in a time-dependent manner. However, maximum phosphorylations of Smad2 by PGD2 and TGF-β1 were achieved at 60 min and 5 min, respectively. To investigate the involvement of TGF-β1 in the PGD2-induced EMT, we examined the effect of TGF-β1 knockdown on the PGD2-induced EMT. As shown in Fig. 3D and 3E, silencing of TGF-β1 significantly suppressed the expression of TGF-β1 mRNA. In the same context, PGD2-induced phosphorylation of Smad2 (Fig. 3F) and EMT (Fig. 3G, 3H and 3I) were significantly suppressed by the silencing of TGF-β1.
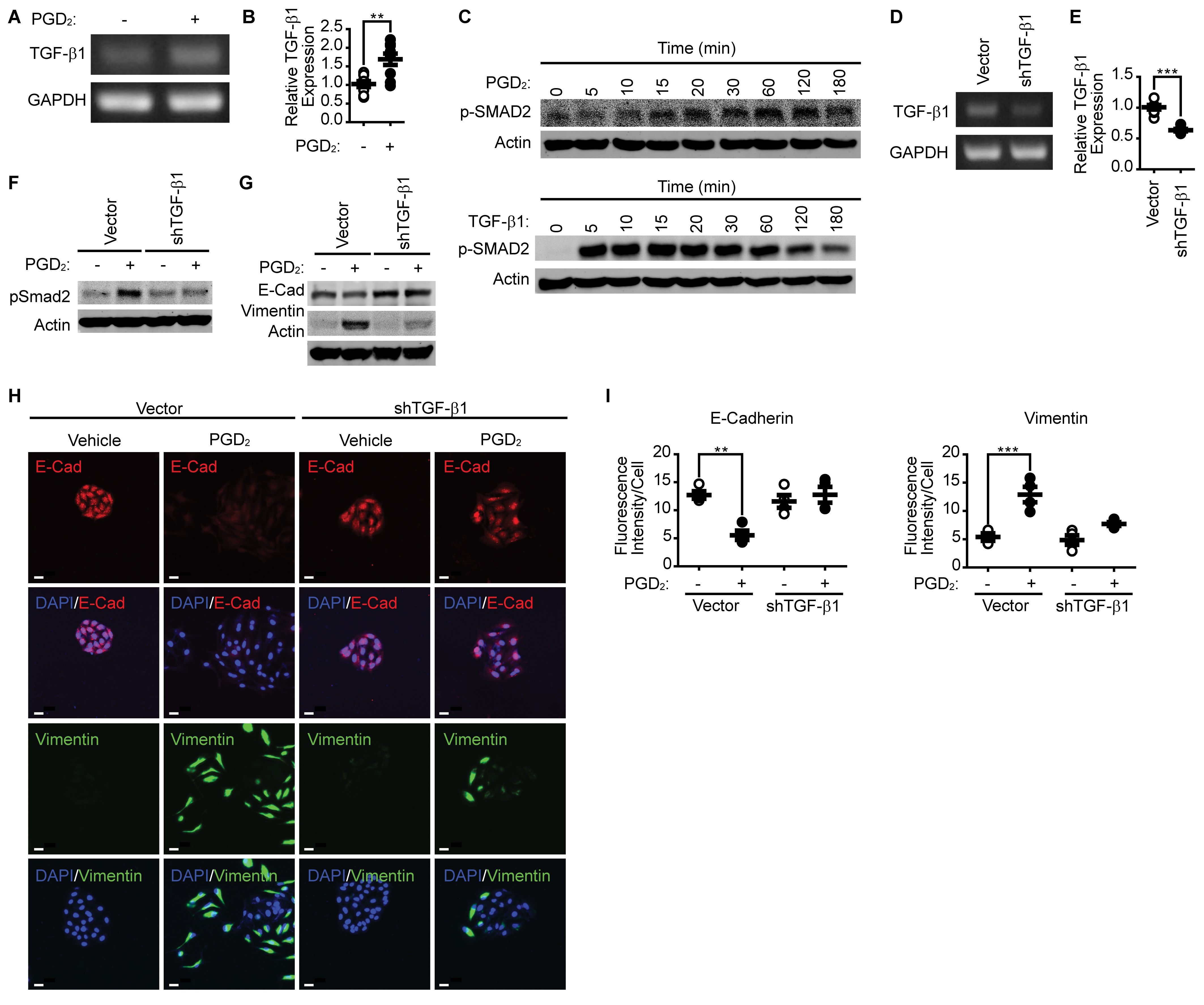
Pharmacological inhibition of TGFβR1 suppresses PGD 2 -induced EMT
Since silencing of TGF-β1 suppressed PGD2-induced Smad2 phosphorylation and EMT, we examined the effect of TGFβR1 inhibition on the PGD2-induced Smad2 phosphorylation and EMT. As shown in Fig. 4A, pharmacological inhibition of TGFβR1 by SB431542 significantly suppressed both TGF-β1- and PGD2-induced Smad2 phosphorylation. In addition, inhibition of TGFβR1 suppressed downregulation of E-cadherin and upregulation of Vimentin (Fig. 4B). Similar results were obtained by immunocytochemical analyses, as shown in Fig. 4C and 4D.
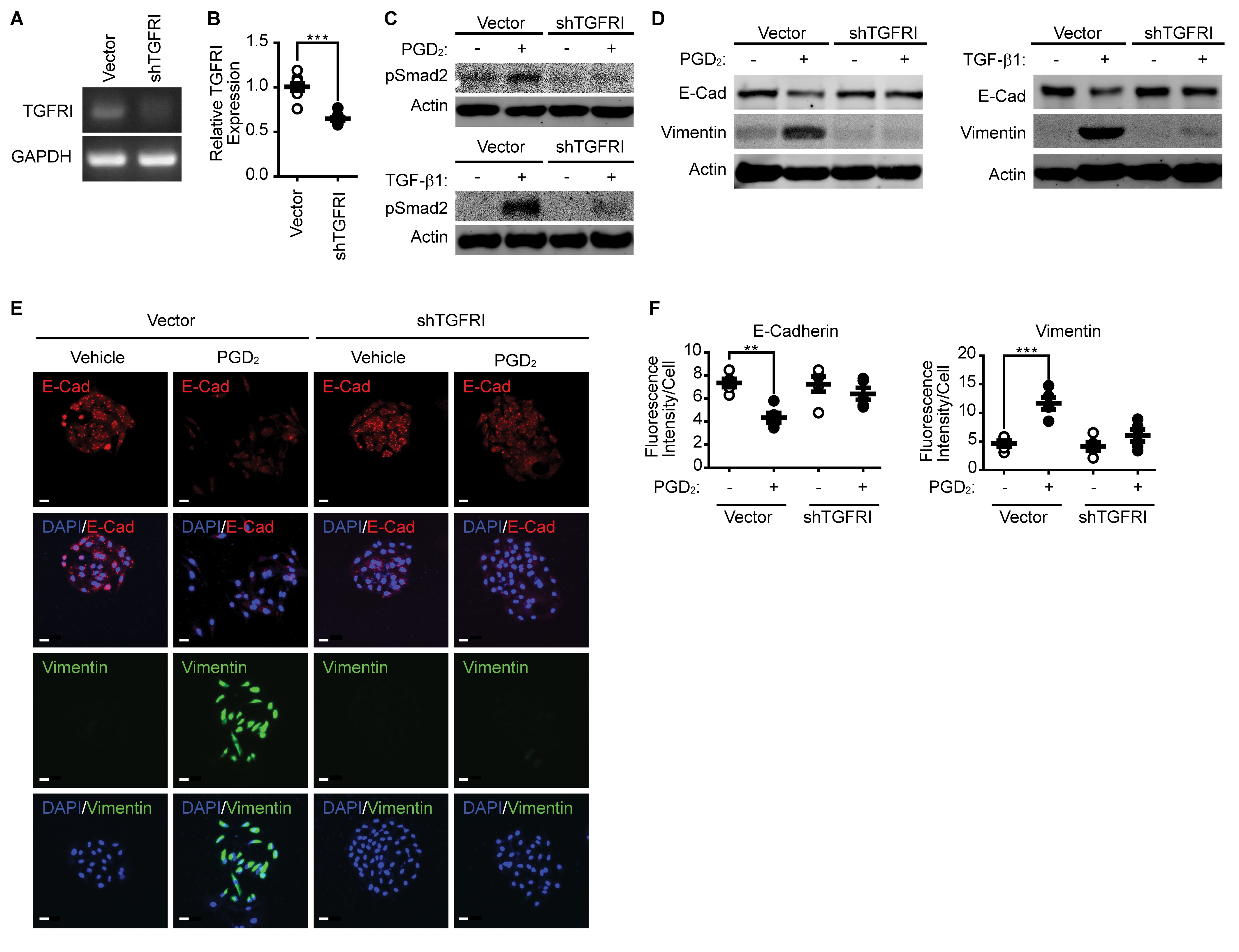
Silencing of TGFβR1 expression suppressed PGD 2 -induced Smad2 phosphorylation and EMT
To further confirm the role of TGFβR1 signaling in PGD2-induced EMT, we examined the effect of TGFβR1 silencing on Smad2 phosphorylation and EMT. As shown in Fig. 5A and 5B, silencing of TGFβR1 significantly reduced mRNA expression of TGFβR1. Silencing of TGFβR1 significantly blocked both TGF-β1 (Fig. 5C, lower panel) and PGD2-induced (Fig. 5C, upper panel) Smad2 phosphorylation. In addition, both TGF-β1 and PGD2-induced EMT were significantly blocked by the silencing of TGFβR1 (Fig. 5D). The effect of TGFβR1 silencing on EMT was further confirmed by the immunocytochemistry results. As shown in Fig. 5E and 5F, silencing of TGFβR1 significantly suppressed downregulation of E-cadherin and upregulation of Vimentin.
Nutrition and serum contents affect PGD 2 -induced EMT
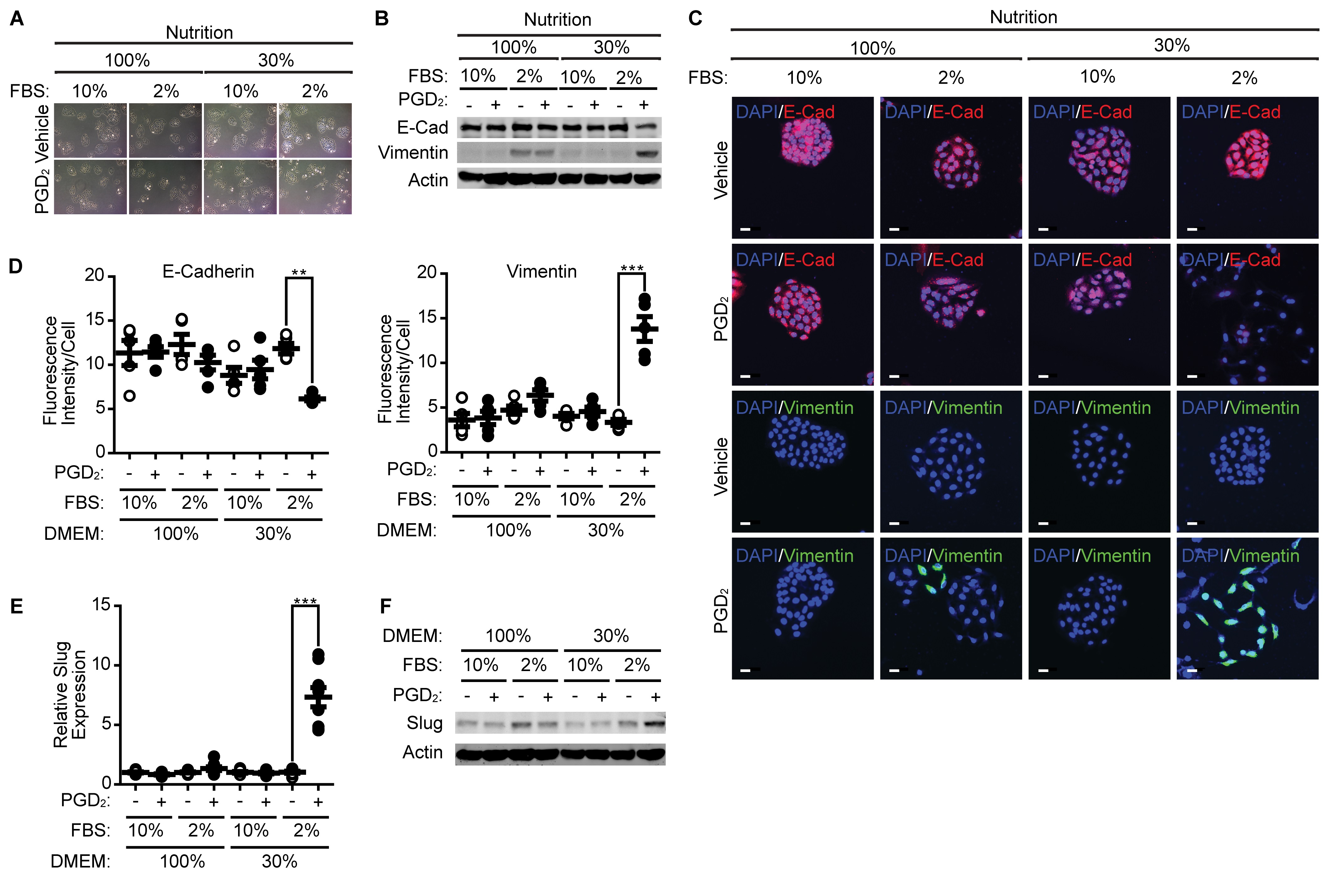
Since nutrition and growth factors, in addition to inflammatory cytokines, are TME factors, we next examined the effect of nutrient and serum levels on EMT. As shown in Fig. 6A, mesenchymal morphology of A549 cells was markedly observed in the presence of PGD2 in low nutrient and low serum conditions. The effect of PGD2 on the expression of E-cadherin and Vimentin was nearly negated in high serum and high nutrient conditions, whereas the effect of PGD2 on the expression of E-cadherin and Vimentin was significantly augmented under low serum and low nutrient conditions (Fig. 6B). Similar results were obtained by immunocytochemical analysis (Fig. 6C and 6D). In addition, Slug expression was markedly enhanced by PGD2 under low serum and low nutrient conditions (Fig. 6E and 6F).
Mesenchymal-Epithelial Transition (MET) is induced at normal culture condition
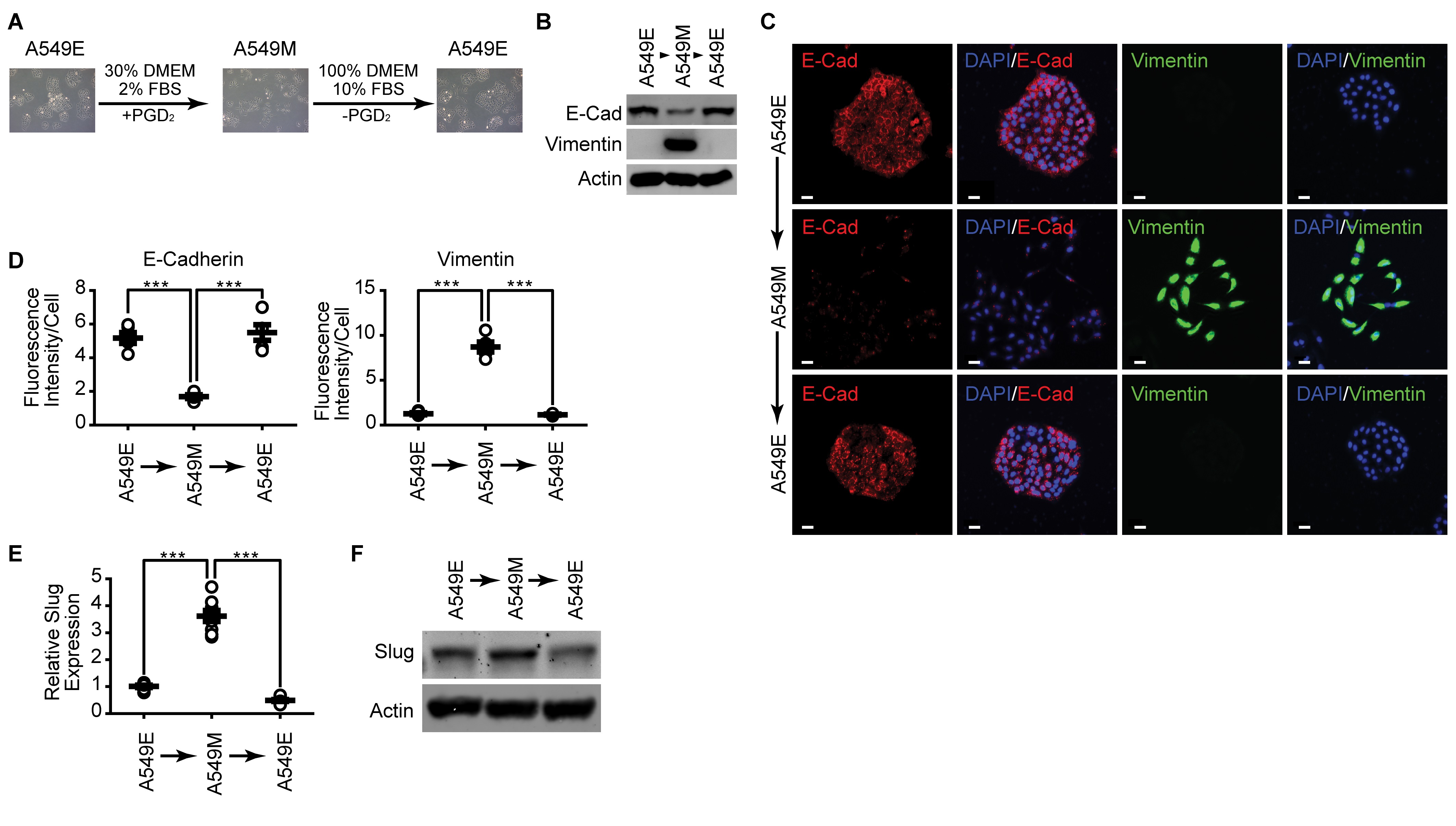
Since our previous results demonstrated that TME factors such as inflammatory cytokines, low nutrient, and low growth factor concentrations could affect EMT, we next examined whether the mesenchymal properties of A549 cells could be perpetuated in normal culture conditions. As shown in Fig. 7A, the mesenchymal type of morphology (A549M) was manifested in the epithelial type cells (A549E) cells when cultured under a normal culture condition. In addition, culture of A549M cells under a normal culture condition reverted the expression level of E-cadherin and Vimentin to that of the initial epithelial stage (Fig. 7B). Similar results were obtained by immunocytochemical analysis (Fig. 7C and 7D). Moreover, the enhanced expression level of Slug in A549M cells was downregulated when the cells are cultured in normal culture conditions (Fig. 7E and 7F).
Author Contributions
Performance of experiments and analysis of data were done by Farzaneh Vafaeinik. Hye Jin Kum and Seo Yeon Jin supported the experiments. Do Sik Min, Sang Heon Song, Hong Koo Ha, Chi Dae Kim discussed this project. Sun Sik Bae generated the idea and wrote the manuscript.
Funding
This work was supported in part by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2020R1A2C2008870, 2017R1A2B4002249).
Statement of Ethics
The authors have no ethical conflicts to disclose.
The authors declare that no conflicts of interests exist.
| 1 Maurya DK, Nandakumar N, Devasagayam TP: Anticancer property of gallic acid in A549, a human lung adenocarcinoma cell line, and possible mechanisms. J Clin Biochem Nutr 2011;48:85-90. https://doi.org/10.3164/jcbn.11-004FR |
||||
| 2 Yao YH, Cui Y, Qiu XN, Zhang LZ, Zhang W, Li H, Yu JM: Attenuated LKB1-SIK1 signaling promotes epithelial-mesenchymal transition and radioresistance of non-small cell lung cancer cells. Chin J Cancer 2016;35:50. https://doi.org/10.1186/s40880-016-0113-3 |
||||
| 3 Risolino M, Mandia N, Iavarone F, Dardaei L, Longobardi E, Fernandez S, Talotta F, Bianchi F, Pisati F, Spaggiari L, Harter PN, Mittelbronn M, Schulte D, Incoronato M, Di Fiore PP, Blasi F, Verde P: Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc Natl Acad Sci U S A 2014;111:E3775-E3784. https://doi.org/10.1073/pnas.1407074111 |
||||
| 4 Zhu X, Chen L, Liu L, Niu X: EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front Oncol 2019;9:1044. https://doi.org/10.3389/fonc.2019.01044 |
||||
| 5 Ma M, He M, Jiang Q, Yan Y, Guan S, Zhang J, Yu Z, Chen Q, Sun M, Yao W, Zhao H, Jin F, Wei M: MiR-487a Promotes TGF-beta1-induced EMT, the Migration and Invasion of Breast Cancer Cells by Directly Targeting MAGI2. Int J Biol Sci 2016;12:397-408. https://doi.org/10.7150/ijbs.13475 |
||||
| 6 Deng QD, Lei XP, Zhong YH, Chen MS, Ke YY, Li Z, Chen J, Huang LJ, Zhang Y, Liang L, Lin ZX, Liu Q, Li SP, Yu XY: Triptolide suppresses the growth and metastasis of non-small ce-ll lung cancer by inhibiting beta-catenin-mediated epithelial-mesenchymal transition. Acta Pharmacol Sin 2021;42:1486-1497. https://doi.org/10.1038/s41401-021-00657-w |
||||
| 7 Park GB, Kim D, Kim YS, Kim JW, Sun H, Roh KH, Yang JW, Hur DY: Regulation of ADAM10 and ADAM17 by Sorafenib Inhibits Epithelial-to-Mesenchymal Transition in Epstein-Barr Virus-Infected Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci 2015;56:5162-5173. https://doi.org/10.1167/iovs.14-16058 |
||||
| 8 Cai G, Wu D, Wang Z, Xu Z, Wong KB, Ng CF, Chan FL, Yu S: Collapsin response mediator protein-1 (CRMP1) acts as an invasion and metastasis suppressor of prostate cancer via its suppression of epithelial-mesenchymal transition and remodeling of actin cytoskeleton organization. Oncogene 2017;36:546-558. https://doi.org/10.1038/onc.2016.227 |
||||
| 9 Kalluri R: EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest 2009;119:1417-1419. https://doi.org/10.1172/JCI39675 |
||||
| 10 Jung HY, Fattet L, Yang J: Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res 2015;21:962-968. https://doi.org/10.1158/1078-0432.CCR-13-3173 |
||||
| 11 Comito G, Ippolito L, Chiarugi P, Cirri P: Nutritional Exchanges Within Tumor Microenvironment: Impact for Cancer Aggressiveness. Front Oncol 2020;10:396. https://doi.org/10.3389/fonc.2020.00396 |
||||
| 12 Lane AN, Higashi RM, Fan TW: Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes Dis 2020;7:185-198. https://doi.org/10.1016/j.gendis.2019.10.007 |
||||
| 13 Burgos-Panadero R, Lucantoni F, Gamero-Sandemetrio E, Cruz-Merino L, Alvaro T, Noguera R: The tumour microenvironment as an integrated framework to understand cancer biology. Cancer Lett 2019;461:112-122. https://doi.org/10.1016/j.canlet.2019.07.010 |
||||
| 14 Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP: Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J 2004;23:1155-1165. https://doi.org/10.1038/sj.emboj.7600069 |
||||
| 15 Muthusamy BP, Budi EH, Katsuno Y, Lee MK, Smith SM, Mirza AM, Akhurst RJ, Derynck R: ShcA Protects against Epithelial-Mesenchymal Transition through Compartmentalized Inhibition of TGF-beta-Induced Smad Activation. PLoS Biol 2015;13:e1002325. https://doi.org/10.1371/journal.pbio.1002325 |
||||
| 16 Buchanan FG, Wang D, Bargiacchi F, DuBois RN: Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem 2003;278:35451-35457. https://doi.org/10.1074/jbc.M302474200 |
||||
| 17 Murata T, Aritake K, Matsumoto S, Kamauchi S, Nakagawa T, Hori M, Momotani E, Urade Y, Ozaki H: Prostagladin D2 is a mast cell-derived antiangiogenic factor in lung carcinoma. Proc Natl Acad Sci U S A 2011;108:19802-19807. https://doi.org/10.1073/pnas.1110011108 |
||||
| 18 Nagoshi H, Uehara Y, Kanai F, Maeda S, Ogura T, Goto A, Toyo-oka T, Esumi H, Shimizu T, Omata M: Prostaglandin D2 inhibits inducible nitric oxide synthase expression in rat vascular smooth muscle cells. Circ Res 1998;82:204-209. https://doi.org/10.1161/01.RES.82.2.204 |
||||
| 19 Kostenis E, Ulven T: Emerging roles of DP and CRTH2 in allergic inflammation. Trends Mol Med 2006;12:148-158. https://doi.org/10.1016/j.molmed.2006.02.005 |
||||
| 20 Qu WM, Huang ZL, Xu XH, Aritake K, Eguchi N, Nambu F, Narumiya S, Urade Y, Hayaishi O: Lipocalin-type prostaglandin D synthase produces prostaglandin D2 involved in regulation of physiological sleep. Proc Natl Acad Sci U S A 2006;103:17949-17954. https://doi.org/10.1073/pnas.0608581103 |
||||
| 21 Maher SA, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Bonvini SJ, Grace MS, Belvisi MG: Prostaglandin D2 and the role of the DP1, DP2 and TP receptors in the control of airway reflex events. Eur Respir J 2015;45:1108-1118. https://doi.org/10.1183/09031936.00061614 |
||||
| 22 Tanaka K, Ogawa K, Sugamura K, Nakamura M, Takano S, Nagata K: Cutting edge: differential production of prostaglandin D2 by human helper T cell subsets. J Immunol 2000;164:2277-2280. https://doi.org/10.4049/jimmunol.164.5.2277 |
||||
| 23 Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K: Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 2001;193:255-261. https://doi.org/10.1084/jem.193.2.255 |
||||
| 24 Menter DG, Dubois RN: Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol 2012;2012:723419. https://doi.org/10.1155/2012/723419 |
||||
| 25 Amano H, Hayashi I, Endo H, Kitasato H, Yamashina S, Maruyama T, Kobayashi M, Satoh K, Narita M, Sugimoto Y, Murata T, Yoshimura H, Narumiya S, Majima M: Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J Exp Med 2003;197:221-232. https://doi.org/10.1084/jem.20021408 |
||||
| 26 Shao J, Sheng GG, Mifflin RC, Powell DW, Sheng H: Roles of myofibroblasts in prostaglandin E2-stimulated intestinal epithelial proliferation and angiogenesis. Cancer Res 2006;66:846-855. https://doi.org/10.1158/0008-5472.CAN-05-2606 |
||||
| 27 Pradono P, Tazawa R, Maemondo M, Tanaka M, Usui K, Saijo Y, Hagiwara K, Nukiwa T: Gene transfer of thromboxane A(2) synthase and prostaglandin I(2) synthase antithetically altered tumor angiogenesis and tumor growth. Cancer Res 2002;62:63-66. | ||||
| 28 Zhang A, Dong Z, Yang T: Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am J Physiol Renal Physiol 2006;291:F1332-1342. https://doi.org/10.1152/ajprenal.00131.2006 |
||||
| 29 Zhang A, Wang MH, Dong Z, Yang T: Prostaglandin E2 is a potent inhibitor of epithelial-to-mesenchymal transition: interaction with hepatocyte growth factor. Am J Physiol Renal Physiol 2006;291:F1323-1331. https://doi.org/10.1152/ajprenal.00480.2005 |
||||
| 30 Jung J, Lee YJ, Choi YH, Park EM, Kim HS, Kang JL: Gas6 Prevents Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells via Production of PGE(2), PGD(2) and Their Receptors. Cells 2019;8:643. https://doi.org/10.3390/cells8070643 |
||||
| 31 Labrecque P, Roy SJ, Fréchette L, Iorio-Morin C, Gallant MA, Parent JL: Inverse agonist and pharmacochaperone properties of MK-0524 on the prostanoid DP1 receptor. PLoS One 2013;8:e65767. https://doi.org/10.1371/journal.pone.0065767 |
||||
| 32 Moon TC, Campos-Alberto E, Yoshimura T, Bredo G, Rieger AM, Puttagunta L, Barreda DR, Befus AD, Cameron L: Expression of DP2 (CRTh2), a prostaglandin D₂ receptor, in human mast cells. PLoS One 2014;9:e108595. https://doi.org/10.1371/journal.pone.0108595 |
||||
| 33 Evdokimova V, Tognon C, Ng T, Sorensen PH: Reduced proliferation and enhanced migration: two sides of the same coin? Molecular mechanisms of metastatic progression by YB-1. Cell Cycle 2009;8:2901-2906. https://doi.org/10.4161/cc.8.18.9537 |
||||
| 34 Shin S, Buel GR, Nagiec MJ, Han MJ, Roux PP, Blenis J, Yoon SO: ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc Natl Acad Sci U S A 2019;116:2967-2976. https://doi.org/10.1073/pnas.1811923116 |
||||
| 35 Li L, Qi L, Liang Z, Song W, Liu Y, Wang Y, Sun B, Zhang B, Cao W: Transforming growth factor-beta1 induces EMT by the transactivation of epidermal growth factor signaling through HA/CD44 in lung and breast cancer cells. Int J Mol Med 2015;36:113-122. https://doi.org/10.3892/ijmm.2015.2222 |
||||
| 36 Choi J, Suh JY, Kim DH, Na HK, Surh YJ: 15-Deoxy-Delta(12,14)-prostaglandin J2 Induces Epithelial-to-mesenchymal Transition in Human Breast Cancer Cells and Promotes Fibroblast Activation. J Cancer Prev 2020;25:152-163. https://doi.org/10.15430/JCP.2020.25.3.152 |
||||
| 37 Medici D, Hay ED, Olsen BR: Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 2008;19:4875-4887. https://doi.org/10.1091/mbc.e08-05-0506 |
||||
| 38 Liu S, Shi L, Wang Y, Ye D, Ju H, Ma H, Yang W, Wang Y, Hu J, Deng J, Zhang Z: Stabilization of Slug by NF-kappaB is Essential for TNF-alpha -Induced Migration and Epithelial-Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma Cells. Cell Physiol Biochem 2018;47:567-578. https://doi.org/10.1159/000489990 |
||||
| 39 Yeh HW, Hsu EC, Lee SS, Lang YD, Lin YC, Chang CY, Lee SY, Gu DL, Shih JH, Ho CM, Chen CF, Chen CT, Tu PH, Cheng CF, Chen RH, Yang RB, Jou YS: PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol 2018;20:479-491. https://doi.org/10.1038/s41556-018-0062-y |
||||
| 40 Xu J, Lamouille S, Derynck R: TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009;19:156-172. https://doi.org/10.1038/cr.2009.5 |
||||
| 41 Jeong KH, Jung JH, Kim JE, Kang H: Prostaglandin D2-Mediated DP2 and AKT Signal Regulate the Activation of Androgen Receptors in Human Dermal Papilla Cells. Int J Mol Sci 2018;19:556. https://doi.org/10.3390/ijms19020556 |
||||
| 42 Hou X, Arvisais EW, Jiang C, Chen DB, Roy SK, Pate JL, Hansen TR, Rueda BR, Davis JS: Prostaglandin F2alpha stimulates the expression and secretion of transforming growth factor B1 via induction of the early growth response 1 gene (EGR1) in the bovine corpus luteum. Mol Endocrinol 2008;22:403-414. https://doi.org/10.1210/me.2007-0272 |
||||
| 43 Jimenez-Segovia A, Mota A, Rojo-Sebastian A, Barrocal B, Rynne-Vidal A, Garcia-Bermejo ML, Gomez-Bris R, Hawinkels L, Sandoval P, Garcia-Escudero R, Lopez-Cabrera M, Moreno-Bueno G, Fresno M, Stamatakis K: Prostaglandin F2alpha-induced Prostate Transmembrane Protein, Androgen Induced 1 mediates ovarian cancer progression increasing epithelial plasticity. Neoplasia (New York, NY) 2019;21:1073-1084. https://doi.org/10.1016/j.neo.2019.10.001 |
||||
| 44 Chen Q, Yang W, Wang X, Li X, Qi S, Zhang Y, Gao MQ: TGF-beta1 Induces EMT in Bovine Mammary Epithelial Cells Through the TGFbeta1/Smad Signaling Pathway. Cell Physiol Biochem 2017;43:82-93. https://doi.org/10.1159/000480321 |
||||
| 45 Chen SH, Sung YF, Oyarzabal EA, Tan YM, Leonard J, Guo M, Li S, Wang Q, Chu CH, Chen SL, Lu RB, Hong JS: Physiological Concentration of Prostaglandin E(2) Exerts Anti-inflammatory Effects by Inhibiting Microglial Production of Superoxide Through a Novel Pathway. Mol Neurobiol 2018;55:8001-8013. https://doi.org/10.1007/s12035-018-0965-4 |
||||
| 46 Han L, Luo H, Huang W, Zhang J, Wu D, Wang J, Pi J, Liu C, Qu X, Liu H, Qin X, Xiang Y: Modulation of the EMT/MET Process by E-Cadherin in Airway Epithelia Stress Injury. Biomolecules 2021;11:669. https://doi.org/10.3390/biom11050669 |
||||
| 47 Chiba T, Ishisaki A, Kyakumoto S, Shibata T, Yamada H, Kamo M: Transforming growth factor-beta1 suppresses bone morphogenetic protein-2-induced mesenchymal-epithelial transition in HSC-4 human oral squamous cell carcinoma cells via Smad1/5/9 pathway suppression. Oncol Rep 2017;37:713-720. https://doi.org/10.3892/or.2016.5338 |
||||
| 48 Mesquita-Santos FP, Bakker-Abreu I, Luna-Gomes T, Bozza PT, Diaz BL, Bandeira-Melo C: Co-operative signalling through DP(1) and DP(2) prostanoid receptors is required to enhance leukotriene C(4) synthesis induced by prostaglandin D(2) in eosinophils. Br J Pharmacol 2011;162:1674-1685. https://doi.org/10.1111/j.1476-5381.2010.01086.x |
||||