×
![]()
Corresponding Author: Suhail Al-Salam
Department of Pathology, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain PO Box 17666 (UAE)
Tel. +97137137464, Fax +97137671966, E-Mail suhaila@uaeu.ac.ae
Galectin-3 Mitigates Cardiomyocytes Injury through Modulation of Left Ventricular Cathepsins B, D, L and S at 24-Hour Post Myocardial Infarction
Suhail Al-Salama Satwat Hashmib Govindan Jagadeesha
aDepartment of Pathology, College of Medicine and Health Sciences, United Arab Emirates University, Al-Ain, United Arab Emirates, bDepartment of Biological and Biomedical Sciences, Aga Khan University, Karachi, Pakistan
Introduction
Myocardial infarction (MI) is a grave medical event that develops following complete occlusion of coronary arteries or their branches. The acute hypoxic event affecting ischemic myocardium will induce a series of complex processes that involve many intracellular signaling pathways leading to structural and contractile dysfunction [1]. Cathepsins are lysosomal proteases that degrade proteins. There are around 15 cathepsins and each of them degrades a different protein, have different structures and work through different mechanisms [2, 3].
The expression of Cathepsins genes was reported to be increased in cardiac stress, remodeling, and dysfunction [4]. Cathepsin S was found to be increased during myocardial pressure overload in rodents and humans with hypertension-induced heart failure [4].
In addition, active Cathepsins S, and L can degrade extracellular matrix (ECM) proteins, including laminin [5], fibronectin [6], elastin [7], and collagens [8]. It is well known that matrix metalloproteinases (MMPs) can degrade all ECM proteins and activate cysteine cathepsins [9]. These outcomes support the notion that cathepsins may participate in cardiac remodeling by mediating ECM degradation in cooperation with other proteases such as MMPs and serine proteases.
Furthermore, the levels of the cathepsin B and S genes have been shown to be increased in the hypertrophic and failing myocardium [4].
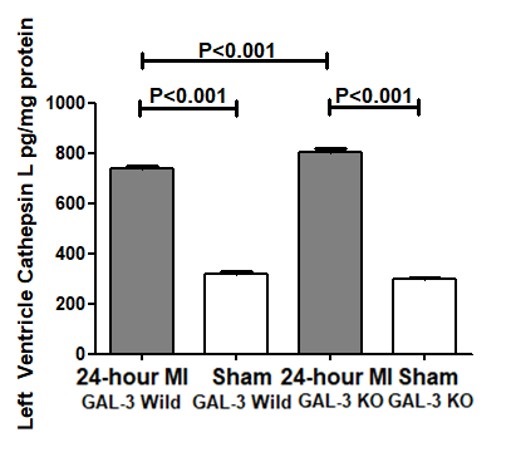
In humans, the levels of cathepsins B, L and S, were increased in patients with dilated and hypertrophic cardiomyopathies compared with normal controls [10].
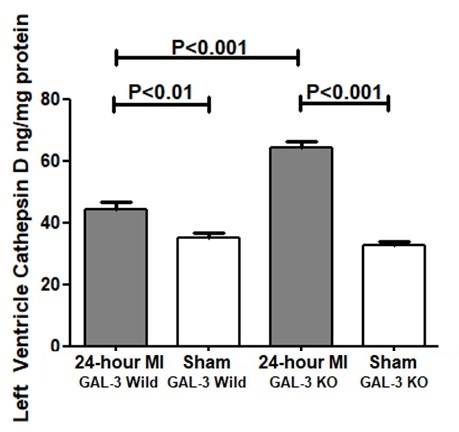
Cathepsin D was found to be elevated in the plasma and hearts of patients following MI [11].
Galectin-3 (GAL-3) is a beta galactoside binding lectin that plays diverse roles in normal and pathological conditions [12]. GAL-3 has been linked with heart failure in recent years [13]. GAL-3 has been shown to coordinate intracellular processes for lysosomal repair and replacement [14].
Our previous results have shown substantial increase in GAL-3 levels in the cardiomyocyes, endothelial cells and neutrophil polymorphs in the heart as well as plasma at 24-hour post MI [15]. We hypothesized that high level of GAL-3 at 24- hour post MI time has a protective role on the heart through modulating myocardial injury which is reflected by lower levels of cathepsins in the myocardium. We used male C57BL6 mice and GAL-3 KO mice with the same background strain to look for cathepsins levels in the left ventricle, in the presence and absence of GAL-3, at 24-hour post MI heart samples.
Materials and Methods
Murine model of myocardial infarction
Male C57B6/J mice (n=16) and GAL-3 KO mice (n=16) were divided into 24-hour MI group and sham operated groups. GAL-3 wild and KO mice (male, age: 12-16 weeks; wt: 20-25g) were anesthetized by an intraperitoneal injection of Phenobarbitone (70mg/kg). The mice were then intubated by transesophageal illumination using a modified 22-gauge plastic cannula and fixed on the operating pad in the supine position by taping all four extremities. The mice were connected to a mouse ventilator (Harvard apparatus Minivent Hugo Sachs Electronik) which supplied room air supplemented with 100% oxygen (tidal volume 0.2 ml/min., rate 120 strokes/min). Rectal temperature was continuously monitored and maintained within 36-37 °C using a heat pad. The lead II ECG (AD Instrument multi-channel recorder interfaced with a computer running Power lab 4/30 data acquisition software) was recorded from needle electrodes inserted subcutaneously. Myocardial infarction was induced in the mice by permanently occluding the left anterior descending coronary artery (LAD) as described earlier [16, 17].
Briefly, the chest was opened with a lateral incision at the 4th intercostal space on the left side of the sternum. Then the chest wall was retracted for better visualization of the heart. With minimal manipulation, the pericardial sac was removed and the left anterior descending artery (LAD) was visualized with a stereomicroscope (Zeiss STEMI SV8). An 8-0 ophthalmic silk suture was passed under the LAD and ligated 1mm distal to left atrial appendage. Occlusion was confirmed by observing immediate paleness of the left ventricle post ligation. An accompanying ECG recording showed characteristic ST-Elevation which further confirmed ischemia. The chest wall was then closed by approximating the third and fourth ribs with one or two interrupted sutures. The muscles were returned back to their original position and the skin closed with 4-0 prolene suture. The animal was gently disconnected from the ventilator and spontaneous breathing was seen immediately. Post-operative analgesic (Butorphanol 2mg/kg, s/c, 6 hourly) was given at the end of the procedure. Sham- operated mice underwent exactly the same procedure described above, except that the suture passed under the LAD is left open and untied. According to the experimental protocol, mice were sacrificed 24 hours after induction of myocardial infarction. The hearts were washed in ice cold PBS, right ventricle and both atria were dissected away and left ventricle was immediately frozen in liquid nitrogen and later stored in -80 °C freezer. Blood was also collected in EDTA vacutainers and centrifuged at 3000 RPM for 15 minutes. The plasma was collected, alliquoted and stored at -80 °C until further analysis.
Protein extraction from samples
Total protein was extracted from heart samples by homogenizing with lysis buffer and collecting the supernatant after centrifugation. For total tissue homogenate, the Left ventricular (LV) heart samples were thawed, weighed and put in cold lysis buffer containing 50mM Tris, 300mM NaCl, 1mM MgCl2, 3mM EDTA, 20mM β-glycerophosphate, 25mM NaF, 1% Triton X-100, 10%w/v Glycerol and protease inhibitor tablet (Roche Complete protease inhibitor cocktail tablets). The hearts were homogenized on ice by a homogenizer (IKA T25 Ultra Turrax). The samples were then centrifuged at 14000 RPM for 15 minutes at 4°C, supernatant collected, alliquoted and stored at -80 °C until further analysis. Total protein concentration was determined by BCA protein assay method (Thermo Scientific Pierce BCA Protein Assay Kit).
Tissue Processing
Hearts were excised, washed with ice-cold phosphate buffer saline (PBS), blotted with filter paper and weighed. Each heart was sectioned into coronal slices of 2mm thickness then cassetted and fixed directly in 10% neutral formalin for 24 hours, which was followed by dehydration in increasing concentrations of ethanol, clearing with xylene and embedding with paraffin.
Immunofluorescent double labeling
Five-um sections were deparaffinized with xylene and rehydrated with graded alcohol. Sections were placed in EnVisionTM FLEX Target Retrieval Solution with a high PH (PH 9) (DAKO Agilent, USA) in a water bath at 95 °C for 30 minutes hour. Sections were washed with distilled water for 5 minutes followed by PBS for 5 minutes. Sections were then incubated with anti-cathepsin D antibody (Rabbit Polyclonal, 1:100, Thermo Fisher Scientific, USA), anti-cathepsin B (Rabbit monoclonal, 1:100, Cell Signaling Technology, USA), anti-cathepsin L (Rabbit monoclonal, 1:100, Abcam, USA), anti-cathepsin S (Rabbit monoclonal, 1:100, Abcam, USA), anti-galectin-3 (Rabbit polyclonal, 1:100, Santa Cruz Biotechnology) overnight at room temperature. Sections were subsequently washed with PBS and incubated with donkey anti-rabbit Rhodamine labeled secondary antibody (Jackson Immune Research Laboratories, USA, 1:100) for one hour at room temperature. Sections were then washed with PBS and incubated with the second primary antibody anti-desmin (Mouse monoclonal, 1:100, DAKO Agilent, USA), overnight at room temperature followed by washing with PBS and incubation with donkey anti-mouse Alexa Fluor 488, (Jackson Immune Research Laboratories, USA, 1:100) antibody for one hour at room temperature. Sections were then washed and mounted in Dapi-water-soluble mounting media (Abcam, USA) and viewed with Olympus Fluorescent microscope. Appropriate positive control sections were used. For negative control, the primary antibody was not added to sections and the whole procedure carried out in the same manner as mentioned above. Positive and negative controls were used in every batch of stained slides (not shown in figures).
Morphometric analysis
Morphometric analysis of GAL-3, cathepsin B, cathepsin D, cathepsin L, and cathepsin S expression in left ventricular cells was done at 24-hour following MI using ImageJ software (http://rsbweb.nih.gov/ij/).
Cathepsin B, cathepsin D, cathepsin L, and cathepsin S labeling was determined by counting the number of positively stained lysosomes within cells in randomly-selected high power fields (HPF) in the left ventricle. The mean numbers of positive lysosomes were converted from per HPF to per mm2 (Each mm2= 4HPF). GAL-3 labeling was determined by counting the number of positively stained cells in randomly-selected high power fields (HPF) in the left ventricle. The mean numbers of positive cells were converted from per HPF to per mm2 (Each mm2= 4HPF).
For, cathepsin B, cathepsin D, cathepsin L, and cathepsin S labeling, cells were considered positive when there was a dotted cytoplasmic staining pattern of lysosomes. For GAL-3 labeling, cells were considered positive when there was a cytoplasmic or nuclear staining pattern or both.
Enzyme linked immunosorbent assay
Left ventricular myocardial concentration of GAL-3, Cathepsin B, Cathepsin D, Cathepsin L, Cathepsin S and Troponin-I were determined using enzyme linked immunosorbent assay (ELISA) development kits: [galectin-3 (DY1197) R&D Systems, Minneapolis, MN, USA], Cathepsin B, (DY2176) R&D Systems, Minneapolis, MN, USA], Cathepsin D, (ab239420) Abcam, Boston, MA, USA], Cathepsin L, (DY952) R&D Systems, Minneapolis, MN, USA], Cathepsin S, (DY1183) R&D Systems, Minneapolis, MN, USA], and cardiac Troponin-I Elisa kit (2010-1-HSP, Life Diagnostics, Inc.), for sandwich ELISA, using standard procedure according to the manufacturer’s instructions. The levels were normalized to total protein concentrations.
Briefly, 96-well plates (Nunc-Immune Plate MaxiSorp Surface (NUNC Brand Products, A/S, Roskilde, Denmark), were coated with antibody specific to our proteins of interest. Biotinylated detection antibody and streptavidin conjugated horseradish peroxidase were used for detection of captured antigens. The plates between steps were aspirated and washed 3 times using ELISA plate washer (BioTek ELx50). Captured proteins were visualized using tetramethylbenzidine (TMB)/hydrogen peroxide. Absorbance readings were made at 450 nm, using a 96-well plate spectrophotometer (BioTek ELx800). Concentrations in the samples were determined by interpolation from a standard curve. Standards and samples were assayed in duplicate.
Statistical analysis
All statistical analyses were done using GraphPad Prism Software version 5. Multiple comparisons between the various groups were achieved by one-way analysis of variance (ANOVA), followed by Newman-Keuls post hoc multiple range tests. Data are presented in mean ± standard error (S.E). P values < 0.05 are considered significant.
Results
Plasma Troponin I at 24- hour post MI
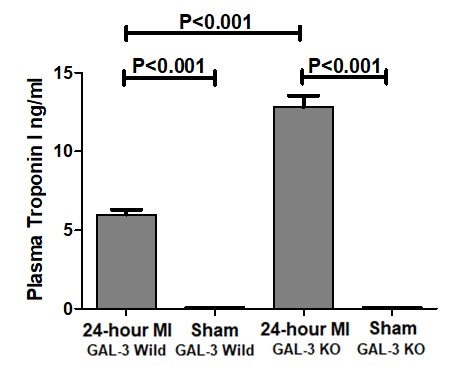
Plasma Troponin I was measured by ELISA at 24-hour post MI in GAL-3 wild and GAL-3 KO MI groups and corresponding sham groups. There were significantly higher levels of plasma Troponin I in both GAL-3 wild (P<0.001) and GAL-3 KO (P<0.001) MI groups as compared to their sham control groups. This confirms acute myocardial infarction in mice in both groups. Moreover, there was a significantly higher levels of plasma Troponin I in GAL-3 KO MI group (12.83 ± 0.7312 ng/ml) than GAL-3 wild MI group (5.992 ± 0.312 ng/ml), (P<0.001), (Fig. 1). This indicates absence of GAL-3 is associated with more damage to the cardiac myocytes and suggests a protective role of GAL-3 at 24-hour post MI.
Galectin-3 is increased in the LV at 24-hour post MI
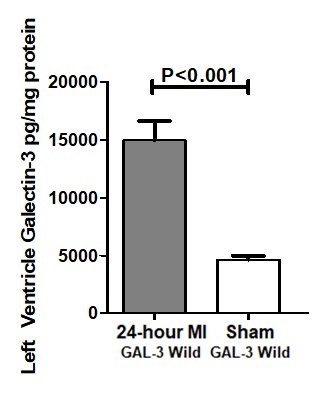
Galectin-3 was measured by ELISA in tissue homogenate of LV at 24-hour post MI hearts in the GAL-3 wild type mice. Our results show GAL-3 level is significantly higher in GAL-3 wild MI group than GAL-3 wild sham group (14950 ± 1662 vs 4628 ± 345.1 pg/mg, P<0.0001), (Fig. 2).
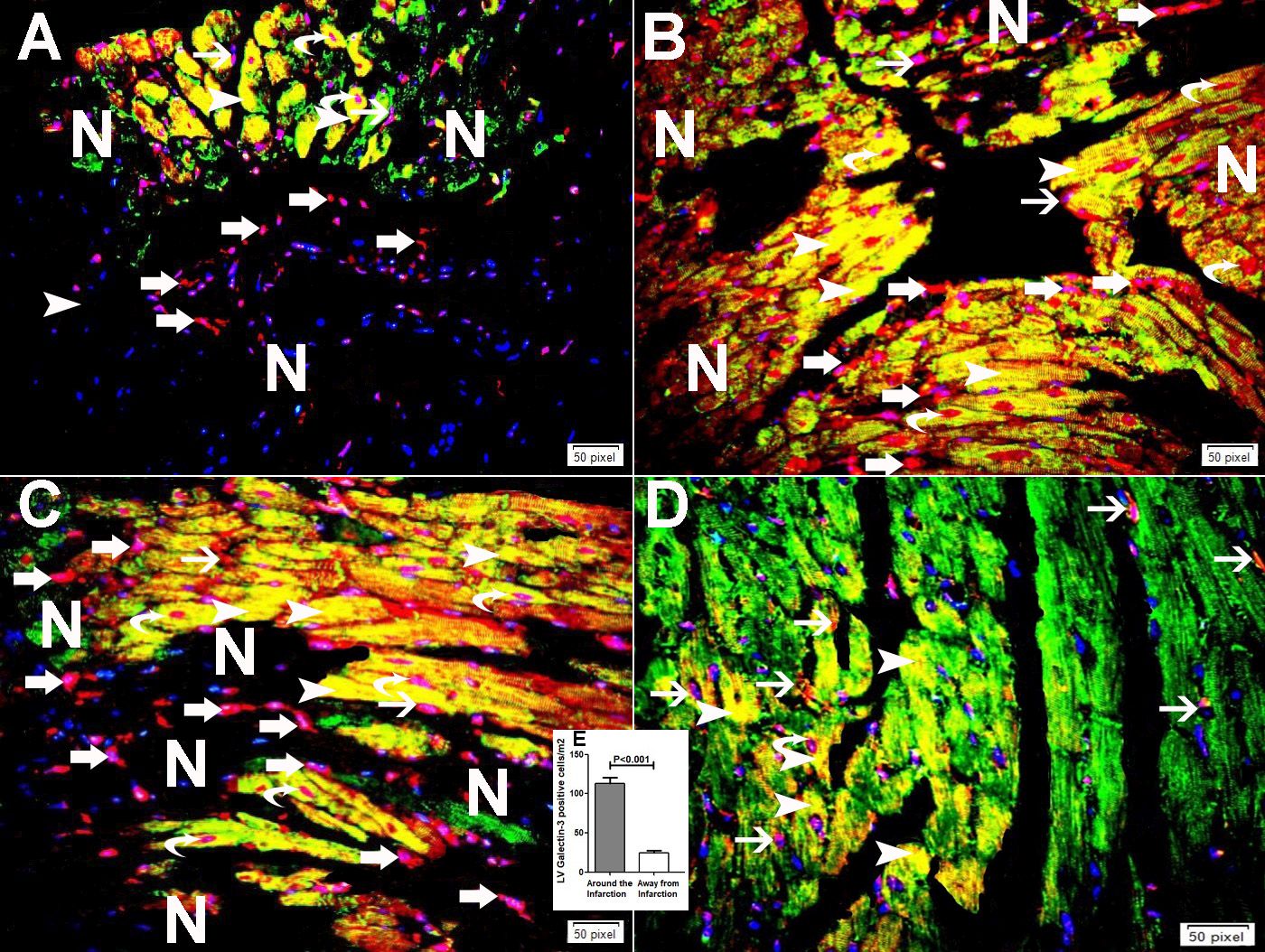
Immunofluorescent double labeling has shown higher expression of GAL-3 in cardiomyocytes, endothelial cell and infiltrating neutrophil polymorphs in areas around the infarction (Fig. 3A-C) when compared to the expression of GAL-3 in areas away from the infarction (Fig. 3D and E).
LV Cathepsins at 24-hour post MI
Cathepsin B Activity. Cathepsin B activity was measured by ELISA in the tissue homogenate of LV of 24- hour post MI hearts in GAL-3 wild and GAL-3 KO MI groups and corresponding sham groups.
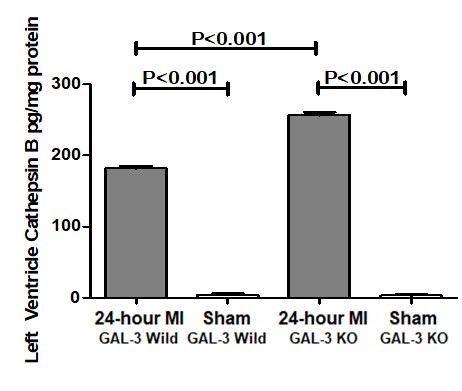
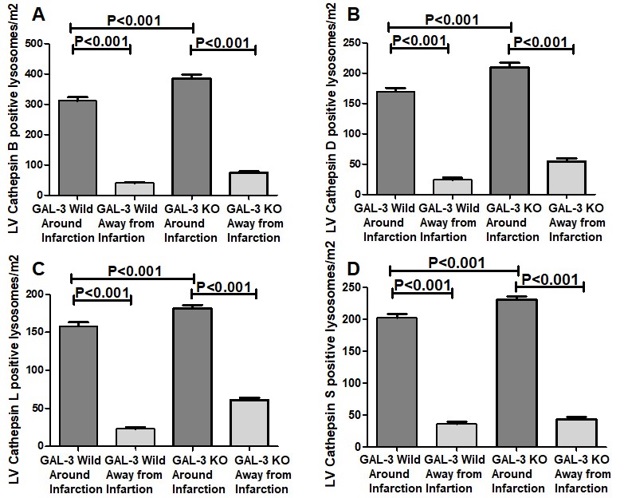
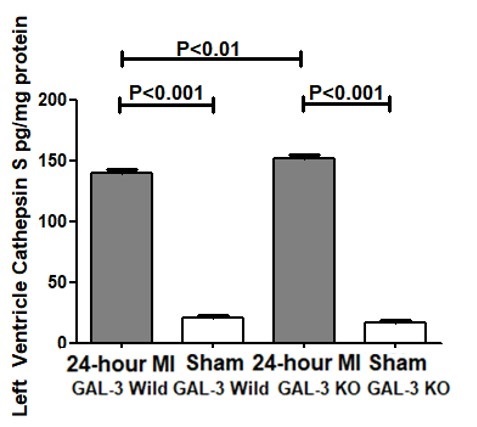
There were significantly higher levels of cathepsin B protein in both GAL-3 wild (P<0.001) and GAL-3 KO (P<0.001) MI groups as compared to their sham control groups. Moreover, there was a significantly higher levels of cathepsin B in GAL-3 KO MI group (256.6 ± 4.192 pg/mg) than GAL-3 wild MI group (181.9 ± 2.392 pg/mg), (P<0.001), (Fig. 4).
Immunofluorescent double labeling has shown increase in the expression of cathepsin B in cardiomyocytes and endothelial cells at 24-hour following MI both in GAL-3 wild and KO MI groups (Fig. 5A and C and Fig. 6A). The pattern of staining was dotted lysosomal staining. There was a significantly higher expression of cathepsin B in areas around infarction than areas away from the infarction in both GAL-3 wild and KO MI groups (Fig. 5A-D and Fig. 6A). We also show a significantly higher expression of cathepsin B in areas around infarction in GAL-3 KO than GAL-3 wild MI groups (Fig. 5A-D and Fig. 6A).
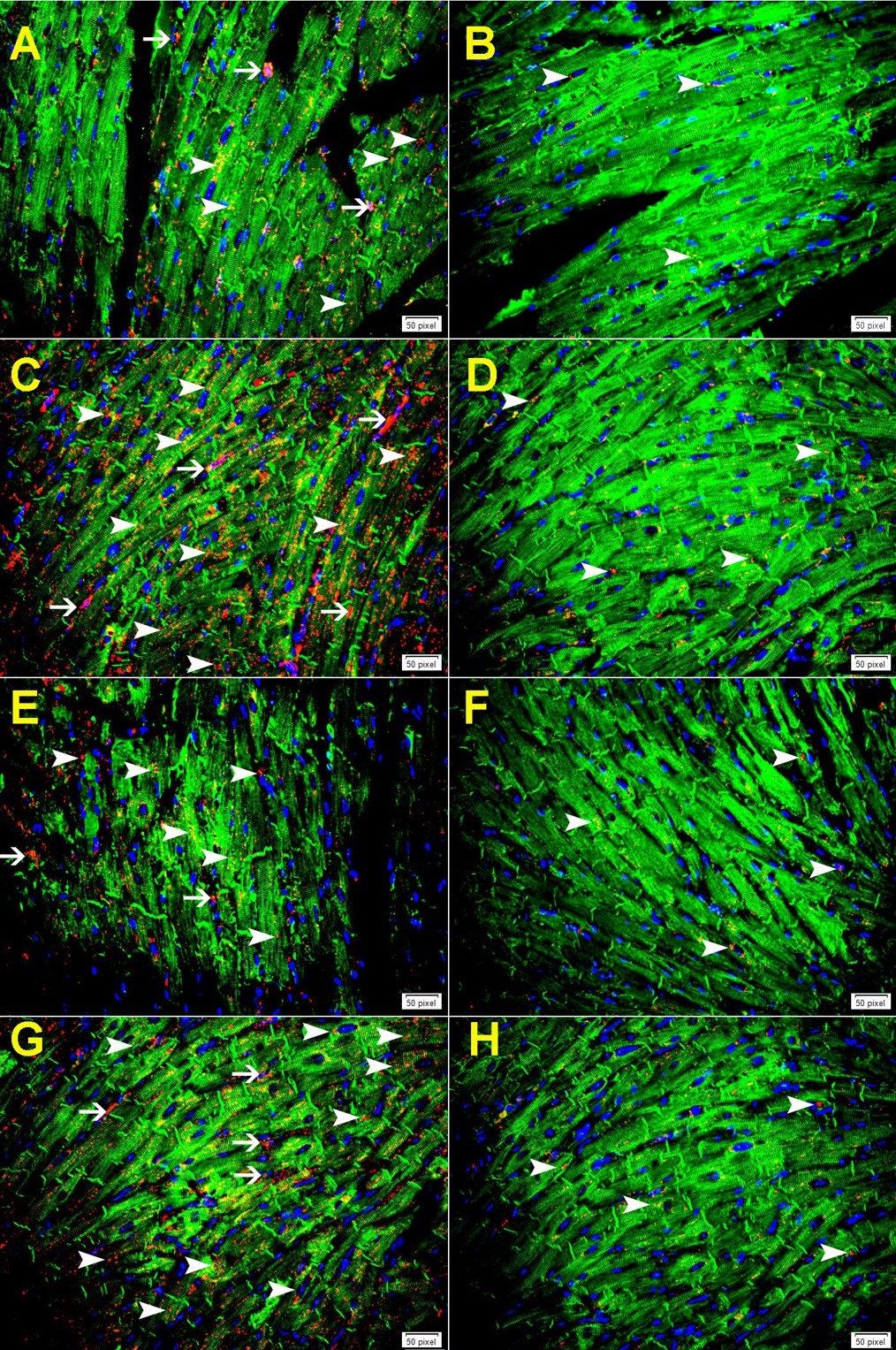
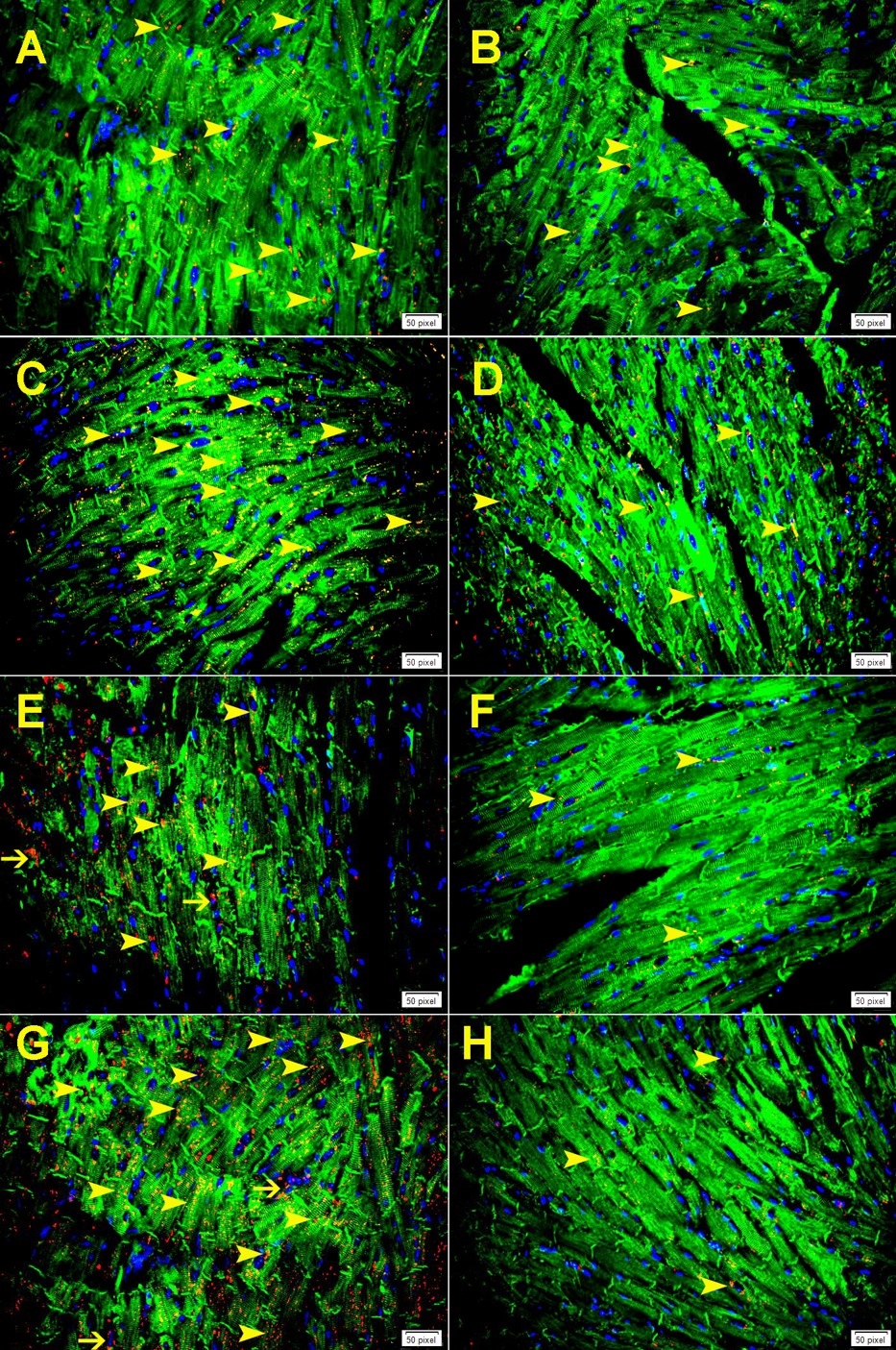
Numerous studies have been done so far linking cathepsins to cardiovascular diseases. Patients with coronary artery stenosis have been shown to have elevated serum cathepsin L levels compared to those without lesions detectable by quantitative angiography [24]. Cathepsin D is shown by proteomic analysis to be increased both in plasma and monocytes of patients with acute coronary syndrome [25]. Elevated levels of cathepsin B is observed in patients in the acute phase of myocardial infarction who have had no past history of ischemic heart disease nor were they on any medication for the same [26]. Studies on the development of atherosclerosis in apolipoprotein E-deficient (ApoE−/−) mice have shown expression of cathepsins B, D and L in macrophages- derived foam cells in the necrotic area of the plaques and fibrous cap of the atherosclerotic lesions [27].
Our results have shown that cathepsins B, D, L, and S are increased in the LV after 24 hours of MI as compared to the sham operated LVs. Immunofluorescent double labeling has also shown that there is significantly higher expression of cathepsins B, D, L, and S in areas around infarction than in areas away from the infarction. Moreover, we have shown that cathepsins B, D, L, and S are expressed in cardiomyocytes and endothelial cells by immunofluorescent double labeling.
Myocardial infarction results in ATP depletion in affected cells causing inhibition of cellular pumps, consequently leading to intracellular accumulation of Na+ and H+ ions. Activation of sodium calcium exchanger leads to increased intracellular Ca2+, increased mitochondrial Ca2+, mitochondrial permeability transition pore opening and necrosis. Mitochondrial swelling and rupture causes increased H+ in the cytoplasm and with the inactivation of H+ pumps leads to decrease in lysosomal pH and subsequent over activation of cathepsin proteases [28]. Lysosomal swelling and membrane rupture causes cathepsins to be released into the cytoplasm and the process of digesting of various substrates commences. Finally, the massive inflow of water into the cell by the osmotic imbalance ultimately leads to cell swelling and rupture of the plasma membrane [29].
Cathepsins have also demonstrated their role in apoptosis and autophagy [30]. Cathepsins play an active part in caspase dependent as well as independent cell death pathways. They are also shown to be important regulators of inflammatory cells like neutrophils, T and B lymphocytes [29]. Cathepsins are released from the lysosomes into the cytosol for its function either by lysosomal destabilization or lysosomal membrane permeabilization [30-32], however, the exact molecular mechanism of its release from cardiomyocyes after MI remains unknown.
Experimental evidence reveals that cathepsin D upregulation after seven days post MI contributes to enhanced myocardial autophagy [33]. The mechanism of this effect is linked to the removal of the autophagosomes in the heart [33]. Studies have overwhelmingly shown that this enhanced myocardial macroautophagy confers a protective role to cathepsin D by promoting protein and organelle quality control [34], which in turn protects against post-MI ventricular remodeling and HF progression.
GAL-3 is a member of a family of lectins with affinity for β-galactoside glycoconjugates. It is ubiquitously present in various organisms [35], as well as expressed by many organs of the body. Galectins are known for their extracellular functions; cell adhesion, migration, signaling, inflammation, fibrosis, infection, cardiovascular diseases, and cancer [36, 37].
Previously, our group has shown that GAL-3 at 24- hour post MI is protective against tissue injury by virtue of its antiapoptotic mechanisms in the myocardium [17]. We also identified that the anti-apoptotic mechanisms are likely mediated through interaction of intracellular GAL-3 with cathepsin D proteins among others. Our results in this study also show that Cathepsins D is higher in GAL-3 KO LVs after 24- hour MI when compared to wild type; in addition, we have tested three other Cathepsins, L, S and B and found the same pattern of expression. In the LVs of Sham group of both wild type and KO, the values of cathepsin, B, D, L and S in the LV are comparable, both have significantly lower values as compared to MI groups and also no significant differences between sham groups was observed.
Our results suggest that there is a possible direct or indirect link between GAL-3 signaling pathway and cathepsins since the absence of LV intracellular GAL-3 in GAL-3 KO MI mice leads to a significantly higher levels of cathepsins D, B, L and S than GAL-3 wild MI mice. We have shown a significantly higher levels of GAL-3 in the LV of GAL-3 wild MI group than sham control group. We have previously alluded to the fact the cathepsin D upregulation post MI time frame is related to its role in myocardial autophagy [33]. GAL-3 is also shown to act as a tag for autophagy receptors like tripartite motif containing 16 (TRIM16) [38, 39]; recruiting the autophagy machinery to damaged organelles after they are marked for degradation. Galectins are shown to form puncta on damaged lysosomes [40], and so serve as markers for endolysosomal damage. Newer studies are however, showing that GAL- 3 is doing more than being just a marker for lysosomal damage.
Endosomal sorting complexes required for transport (ESCRT) is shown to play a role in repair of damaged lysosomes and promotes cell survival [41, 42]; this is especially important in the context of maintaining endolysosomal system homeostasis. GAL-3 and ESCRT Component ALIX interact and protect lysosomes from damage, evidenced by experimental approaches to measure lysosomal membrane repair [41, 42] at various time points.
GAL-3 thus switches from interplay with ESCRT, involved in membrane repair [25, 26], to Tripartite Motif Containing 16 (TRIM16) interactions, involved in autophagic removal of damaged lysosomes [38, 43].
Our results show increase in cathepsin B, D, L and S levels in GAL-3 knockout LVs after 24 hours of MI can be explained on the evidence that expressed GAL-3 during early post MI time point recruits, controls, and integrates ESCRT and autophagy pathways [27], resulting in building up a cellular response to endomembrane damage which may include membrane repair of lysosomal membrane and prevents cathepsins from being released; this explanation supports our finding of significantly lower concentrations of LV cathepsins B, D, L and S in GAL-3 wild MI mice than GAL-3 KO MI mice. Hence GAL-3 serves to activate survival mechanisms to prevent further cardiomyocyes damage this is supported by significantly lower levels of plasma troponin I in GAL-3 wild MI group than GAL-3 KO MI group suggesting that absence of intracellular GAL-3 is associated with more damage to the cardiomyocyes. Studies have shown a close correlation between the levels of cardiac troponins and infarct size in acute MI and that the level of plasma troponin is a measure of the magnitude of infarct size [44, 45]. Troponin I is a very specific cardiomyocyte protein and can only be seen in the peripheral blood when there is injury to cardiac myocytes that allows passage of troponin I to the circulation. However, as we do not have the infarct size measurement per se, we think that it is a limitation of this study and will be considered in our future studies.
So, following MI, GAL-3 is likely to orchestrate a response to either cope and repair damage or escalate its response due to accumulation of unsalvageable organelles.
Conclusion
The increased levels of GAL-3 at 24-hour following MI regulates the process of cardiomyocytes injury through modulation of lysosomal cathepsins B, D, L and S.
The authors would like to thank The Zayed Bin Sultan Center for Health Sciences, College of Medicine & Health Sciences, United Arab Emirates University for their support of this project.
The authors would like to thank Ms. Manjusha Sudhadevi in the department of pathology, College of Medicine &Health Sciences for tissue processing.
Author Contributions
All authors reviewed and approved the submitted version of the manuscript. S.A. introduced the concept, designed the study, analyzed and interpreted the data, performed immunofluorescent double labeling, designed the figures, and wrote the manuscript. S.H. performed the experimental model of MI, analyzed and interpret the data, and wrote the manuscript. G.J. performed protein extraction, ELISA technique and submitted resulted data.
Funding
This project is supported by The Zayed Bin Sultan Center for Health Sciences (ZCHS) Grant 31R176, 21RO73 and CMHS grant 12M130.
Statement of Ethics
All animal experimental procedures are approved by the Animal Research Ethics Committee of the UAE University, Protocol ERA_2020_6175.
The authors declare that no conflicts of interest exist.
| 1 Götte MJ, van Rossum AC, Twisk JWR, Kuijer JPA, Marcus JT, Visser CA: Quantification of regional contractile function after infarction: strain analysis superior to wall thickening analysis in discriminating infarct from remote myocardium. J Am Coll Cardiol 2001;37:808-817. https://doi.org/10.1016/S0735-1097(00)01186-4 |
||||
| 2 Turk V, Turk B, Guncar G, Turk D, Kos J: Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv Enzyme Regul 2002;42:285-303. https://doi.org/10.1016/S0065-2571(01)00034-6 |
||||
| 3 Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D: Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 2012;1824:68-88. https://doi.org/10.1016/j.bbapap.2011.10.002 |
||||
| 4 Cheng XW, Obata K, Kuzuya M, Izawa H, Nakamura K, Asai E, Nagasaka T, Saka M, Kimata T, Noda A, Nagata K, Jin H, Shi GP, Iguchi A, Murohara T, Yokota M: Elastolytic cathepsin induction/activation system exists in myocardium and is upregulated in hypertensive heart failure. Hypertension 2006;48:979-987. https://doi.org/10.1161/01.HYP.0000242331.99369.2f |
||||
| 5 Burns-Kurtis CL, Olzinski AR, Needle S, Fox JH, Capper EA, Kelly FM, McQueney MS, Romanic AM: Cathepsin S expression is up-regulated following balloon angioplasty in the hypercholesterolemic rabbit. Cardiovasc Res 2004;62:610-620. https://doi.org/10.1016/j.cardiores.2004.02.002 |
||||
| 6 Taleb S, Cancello R, Clement K, Lacasa D: Cathepsin S promotes human preadipocyte differentiation: possible involvement of fibronectin degradation. Endocrinology 2006;147:4950-4959. https://doi.org/10.1210/en.2006-0386 |
||||
| 7 Shi GP, Sukhova GK, Kuzuya M, Ye Q, Du J, Zhang Y, Pan JH, Lu ML, Cheng XW, Iguchi A, Perrey S, Lee AM, Chapman HA, Libby P: Deficiency of the cysteine protease cathepsin S impairs micro vessel growth. Circ Res 2003;92:493-500. https://doi.org/10.1161/01.RES.0000060485.20318.96 |
||||
| 8 Cheng XW, Kuzuya M, Nakamura K, Di Q, Liu Z, Sasaki T, Kanda S, Jin H, Shi GP, Murohara T, Yokota M, Iguchi A: Localization of cysteine protease, cathepsin S, to the surface of vascular smooth muscle cells by association with integrin alphanubeta3. Am J Pathol 2006;168:685-694. https://doi.org/10.2353/ajpath.2006.050295 |
||||
| 9 van Hinsbergh VW, Engelse MA, Quax PH: Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol 2006;26:716-728. https://doi.org/10.1161/01.ATV.0000209518.58252.17 |
||||
| 10 Ge J, Zhao G, Chen R, Li S, Wang S, Zhang X, Zhuang Y, Du J, Yu X, Li G, Yang Y: Enhanced myocardial cathepsin B expression in patients with dilated cardiomyopathy. Eur J Heart Fail 2006;8:284-289. https://doi.org/10.1016/j.ejheart.2005.09.004 |
||||
| 11 Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A, Takeyama T, Kawaguchi T, Watanabe T, Fujiwara T, Fujiwara H, Seishima M, Minatoguchi S: The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res 2011;91:330-339. https://doi.org/10.1093/cvr/cvr073 |
||||
| 12 Barondes SH, Cooper DN, Gitt MA, Leffler H: Galectins. Structure and function of a large family of animal lectins. J Biol Chem 1994;269:20807-20810. https://doi.org/10.1016/S0021-9258(17)31891-4 |
||||
| 13 Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, Larson MG, Levy D: Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 2012;60:1249-1256. https://doi.org/10.1016/j.jacc.2012.04.053 |
||||
| 14 Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA, Salemi M, Phinney B, Mari M, Reggiori F, Deretic V: Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev Cell 2020;52:69-87.e8. https://doi.org/10.1016/j.devcel.2019.10.025 |
||||
| 15 Hashmi S, Al-Salam S: Galectin-3 is expressed in the myocardium very early post-myocardial infarction. Cardiovasc Pathol 2015;24:213-223. https://doi.org/10.1016/j.carpath.2014.12.001 |
||||
| 16 Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM: Myocardial ischemia and reperfusion: a murine model. Am J Physiol 1995;269:H2147-2154. https://doi.org/10.1152/ajpheart.1995.269.6.H2147 |
||||
| 17 Al-Salam S, Hashmi S, Jagadeesh GS, Tariq S: Galectin-3: A Cardiomyocyte Antiapoptotic Mediator at 24-Hour Post Myocardial Infarction. Cell Physiol Biochem 2020;54:287-302. https://doi.org/10.33594/000000220 |
||||
| 18 Turk B, Turk D, Turk V: Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta 2000;1477:98-111. https://doi.org/10.1016/S0167-4838(99)00263-0 |
||||
| 19 Vizovisek M, Vidak E, Javorsek U, Mikhaylov G, Bratovs A, Turk B: Expert Opin Ther Targets. 2020;24:573-588. https://doi.org/10.1080/14728222.2020.1746765 |
||||
| 20 Goulet B, Baruch A, Moon NS, Poirier M, Sansregret LL, Erickson A, Bogyo M, Nepveu A: A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell 2004;14:207-219. https://doi.org/10.1016/S1097-2765(04)00209-6 |
||||
| 21 Duncan EM, Muratore-Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF, Allis CD: Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 2008;135:284-294. https://doi.org/10.1016/j.cell.2008.09.055 |
||||
| 22 Ceru S, Konjar S, Maher K, Repnik U, Krizaj I, Bencina M, Renko M, Nepveu A, Zerovnik E, Turk B, Kopitar-Jerala N: Stefin B interacts with histones and cathepsin L in the nucleus. J Biol Chem 2010;285:10078-10086. https://doi.org/10.1074/jbc.M109.034793 |
||||
| 23 Lutgens SP, Cleutjens KB, Daemen MJ, Heeneman S: Cathepsin cysteine proteases in cardiovascular disease. FASEB J 2007;21:3029-3041. https://doi.org/10.1096/fj.06-7924com |
||||
| 24 Liu J, Sukhova GK, Yang JT, Sun J, Ma L, Ren A, Xu WH, Fu H, Golganov GM, Hu C, Libby P, Shi GP: Cathepsin L expression and regulation in human abdominal aortic aneurysm, atherosclerosis, and vascular cells. Atherosclerosis 2006;184:302-311. https://doi.org/10.1016/j.atherosclerosis.2005.05.012 |
||||
| 25 Vivanco F, Martin-Ventura JL, Duran MC, Barderas MG, Blanco-Colio L, Darde VM, Ms S, Meilhac O, Michel JB, Tunon J, Egido J: Quest for novel cardiovascular biomarkers by proteomic analysis. J Proteome Res 2005;4:1181-1191. https://doi.org/10.1021/pr0500197 |
||||
| 26 Shalia KK, Mashru MR, Shah VK, Soneji SL, Payannavar S: Levels of cathepsins in acute myocardial infarction. Indian Heart J 2012;64:290-294. https://doi.org/10.1016/S0019-4832(12)60089-3 |
||||
| 27 Jormsjo S, Wuttge DM, Sirsjo A, Whatling C, Hamsten A, Stemme S, Erikkson P: Differential expression of cysteine and aspartic proteases during progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol 2002;161:939-945. https://doi.org/10.1016/S0002-9440(10)64254-X |
||||
| 28 Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M, Criollo A, Nemchenko A, Hill JA, Lavandero S: Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis 2011;2:e244. https://doi.org/10.1038/cddis.2011.130 |
||||
| 29 Conus S, Simon HU: Cathepsins: key modulators of cell death and inflammatory responses. Biochem Pharmacol 2008;76:1374-1382. https://doi.org/10.1016/j.bcp.2008.07.041 |
||||
| 30 Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, Ojcius DM, Jäättelä M, Kroemer G: Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med 2003;197:1323-1334. https://doi.org/10.1084/jem.20021952 |
||||
| 31 Joy B, Sivadasan R, Abraham TE, John M, Sobhan PK, Seervi M, Santhoshkumar TR: Lysosomal destabilization and cathepsin B contributes for cytochrome c release and caspase activation in embelin-induced apoptosis. Mol Carcinog 2010;49:324-336. https://doi.org/10.1002/mc.20599 |
||||
| 32 Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, Buellesbach J, Borner C: Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 2010;17:1167-1178. https://doi.org/10.1038/cdd.2009.214 |
||||
| 33 Wu P, Yuan X, Li F, Zhang J, Zhu W, Wei M, Li J, Wang X: Myocardial Upregulation of Cathepsin D by Ischemic Heart Disease Promotes Autophagic Flux and Protects Against Cardiac Remodeling and Heart Failure. Circ Heart Fail 2017;10:e004044. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004044 |
||||
| 34 Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A, Takeyama T, Kawaguchi T, Watanabe T, Fujiwara T, Fujiwara H, Seishima M, Minatoguchi S: The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res 2011;91:330-339. https://doi.org/10.1093/cvr/cvr073 |
||||
| 35 Vasta GR, Feng C, Gonzalez-Montalban N, Mancini J, Yang L, Abernathy K, Frost G, Palm C: Functions of galectins as 'self/non-self'-recognition and effector factors. Pathog Dis 2017;75:ftx046. https://doi.org/10.1093/femspd/ftx046 |
||||
| 36 Di Lella S, Sundblad V, Cerliani JP, Guardia CM, Estrin DA, Vasta GR, Rabinovich GA: When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry 2011;50:7842-7857. https://doi.org/10.1021/bi201121m |
||||
| 37 Johannes L, Jacob R, Leffler H. Galectins at a glance. J Cell Sci 2018;131:jcs208884. https://doi.org/10.1242/jcs.208884 |
||||
| 38 Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun JA, Johansen T, Deretic V: TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev Cell 2016;39:13-27. https://doi.org/10.1016/j.devcel.2016.08.003 |
||||
| 39 Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F: Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012;482:414-418. https://doi.org/10.1038/nature10744 |
||||
| 40 Aits S, Kricker J, Liu B, Ellegaard AM, Hamalisto S, Tvingsholm S, Corcelle-Termeau E, Hogh S, Farkas T, Jonassen A, Gromova I, Mortensen M, Jäättelä M: Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015;11:1408-1424. https://doi.org/10.1080/15548627.2015.1063871 |
||||
| 41 Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI: Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018;360:eaar5078. https://doi.org/10.1126/science.aar5078 |
||||
| 42 Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, Stenmark H: ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 2018;37:e99753. https://doi.org/10.15252/embj.201899753 |
||||
| 43 Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA, Salemi M, Phinney B, Mari M, Reggiori F, Deretic V: Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev Cell 2020;52:69-87.e8. https://doi.org/10.1016/j.devcel.2019.10.025 |
||||
| 44 Hallén J: Troponin for the estimation of infarct size: what have we learned? Cardiology 2012;121:204-212. https://doi.org/10.1159/000337113 |
||||
| 45 Metzler B, Hammerer-Lercher A, Jehle J, Dietrich H, Pachinger O, Xu Q, Mair J: Plasma cardiac troponin T closely correlates with infarct size in a mouse model of acute myocardial infarction. Clin Chim Acta 2002;325:87-90. https://doi.org/10.1016/S0009-8981(02)00296-6 |
||||