Cancer Pharmacology Division, CSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu 180001 (India)
E-Mail aremqayoom03@gmail.com
Divergent Signaling Pathways May Lead to Convergence in Cancer Therapy –
A Review
Arem Qayuma,b
Syed Mohmad Shahc
Shashank K. Singha,b
aCancer Pharmacology Division, Indian Institute of Integrative Medicine, CSIR, Jammu, India,
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, India,
cSher-e-Kashmir University of Agricultural Sciences & Technology of Kashmir, Srinagar, India
Key Words
Signaling • Receptors • Transmitters • Kinases • Proliferation and differentiation • Extracellular matrix • Cell death • Cell adhesion • Secondary messengers • Immune checkpoint
Abstract
Cancer is a chaos of uncontrolled cell proliferation that has consistently invented new circuitry programs to operate inside the cell machinery. Globally, cancer statistics account for 65% of mortality worldwide, mainly due to the adoption of lifestyle behaviours. In 2020, FDA approved 40 new drugs, out of which 16 (40%) were approved as cancer drugs. Overall, the risk of dying from cancer decreased, but further reductions in cancer death rates can be accelerated by applying existing cancer control knowledge across all the population segments, emphasising those in the lowest socio-economic and other disadvantaged population. Various therapeutic regimes, including low-molecular-weight inhibitors, targeting oncogenic signaling pathways are under development. However, the pitfall of targeted therapies is the quick emergence of acquired drug resistance encumbered with toxic side effects. Several FDA acclaimed therapeutic legacies or biosimilars earmarked signaling pathways of rare diseases (cystic fibrosis, erythropoietic protoporphyria, neuromyelitis optica spectrum disorder, tenosynovial giant cell tumor, sickle cell disease, systemic sclerosis-associated interstitial lung disease, muscular dystrophy), neurological and psychiatric disorders, infectious diseases, heart, lung, circulatory, endocrine diseases, autoimmune conditions, cancers and blood disorders. When cancer progresses, these signals develop specific characteristics that can be targeted for anti-cancer therapy. The designer inhibitors have emerged as novel pharmaceutical interventions that aim to block the pathways in an effort to reverse the abnormal phenotype of the cancer cells. Numerous cell-signaling channels have evolved and invigorated to make off three-dimensional feedback networks. The magnitude of accessible information by pathways occupies curated information as a consortium. To fully appreciate the pivotal roles that signaling cascades play in tumor development, it is necessary to understand the involved signaling cascades in the interaction between cancer cells. The prime endeavour is to canonically curate all signaling pathways involving cell cycle, EGFR, MAPK, GPCR, PI3K/AKT/mTOR, immune checkpoints, nuclear receptors, janus kinase, transcription activators etc., involving the manipulation of genetic and nuclear receptors. Here, we will summarize the vast amount of information describing the signals that mediate crosstalk between cancer cells and the targets related to this crosstalk.
Background
Cellular signaling is a series of reaction(s) in which a group of cells nominate to work together to govern a series of actions, to activate a hormone/growth factor or to bind to a specific protein receptor, ultimately leading to multiplication or death [1]. These signaling molecules and their interaction mechanisms are amenable to external influence via the agonists and/or antagonists and hence are amenable to corrections, alterations or manipulations in cases where their functioning or interactions pose a challenge to holistic welfare of an organism. These external therapies thus guarantee the survival/welfare of an organism by keeping in check the pathways whenever they go awry, unless and until the pathway develops a resistance or escape mechanism to evade the effect of the drug and ensue a chaos as in cancer. The understanding of the evolution of this resistance to various therapies by signaling pathways forms the core and pivot to develop new strategies for treating the catastrophic disease like cancer. The different characteristics have been adapted by cancer cells through the essential cellular transduction of signals induced by many genetic and epigenetic alterations that drives cancer and other systems involving ECM, immune system and blood vessels [2]. Various inhibitors like anti-angiogenic, antibody-based, checkpoint blockers targeting pathways like tyrosine kinase, mTOR, PI3K, histone deacetylase have a significant impact on cancer growth, even though the recurrence rate is high due to the development of drug resistance [3]. The intricacy of the signaling pathways imposes a great challenge for discovering anti-cancer drugs because of the redundancy of pathways controlling proliferation, crosstalk between compensatory pathways and feedback loop mechanisms that cause reactivation of the pathways. Inhibitor studies, conducted on different effectors like B-Raf, EGFR, Abl to treat various cancer like chronic myelogenous leukaemia, breast cancer, melanoma and non-small-cell lung carcinomas, gave significant results initially but developed resistance at later stages [4]. Major known proliferation pathways such as Ras, PI3K-AKT, EGFR, Wnt, Hedgehog, Myc, NF-κB, EZH2, cyclins etc., have got the probability to get mutated in different types of cancer, and most of the approaches for treatment of the disease are based on targeting these pathways [5].
Moreover, the efficacy of these targeted therapies is based on several factors in which tumor microenvironment promotes the stimulation of other existing pathways which act as soil and seed for maintaining malignancy. Further, tumor heterogeneity complicates the sensitivity to inhibitors targeting mutated signaling proteins. There is a need for an in-depth understanding of the mechanism of resistance to interrogate the diverse signaling network in heterogeneous populations of tumor cells [6, 7]. Whereas, chemotherapy involves combinations based on the signaling pathways related to the resistance against various anti-cancer drugs to escalate the selective execution of cancer cell death, the numerous inhibitors of crucial signaling pathways induced resistance showing high toxicity and compensatory mechanisms. Extraction of assimilated public transcriptomic datasets can rationalize the functions of mammalian cellular signaling pathways. Previously discovered drugs described the efforts in encompassing specific knowledge about the small molecule perturbations in targeting signal transduction pathways. Also, it describes the functional interdependencies through a given response via endocrine, paracrine and cytokine signals between the different classes of inhibitors [8]. The omics project like signaling pathways project (SPP) have been intended to decipher the mechanism of drug interactions. As most signaling pathways were found to be overlapping with other pathways exhibiting compensatory mechanisms, the speculation of a complex signaling for a specific mutation in cancer cells is rendered difficult [9]. Due to the remarkable plasticity of signaling pathways for tumor growth and cell proliferation, the attempts for inhibition of one or two signaling pathways did not yield anticipated success in tumor treatment. To circumvent the problem, attempts are being made to study the effects of combinatorial drug therapies simultaneously inhibiting many interlinked and correlated signaling pathways to regress the tumor growth. This review is an insightful source of signaling pathways in intending to develop an innovative approach for anti-cancer mono and combination therapy and intends to discuss the whole cellular ecosystem where cancer originates and/or where it could be cured [10].
Extracellular matrix (ECM)
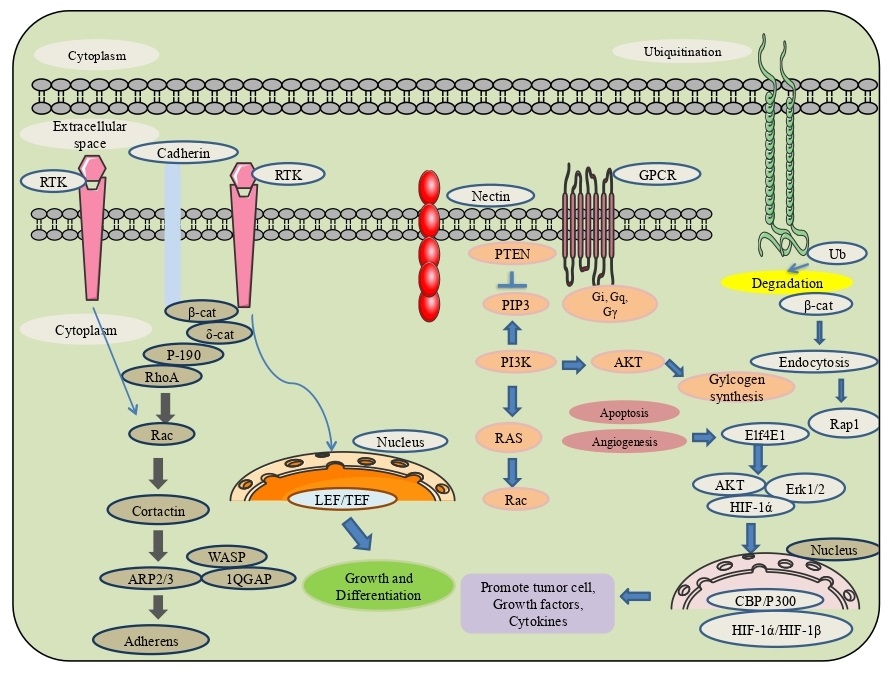
Dynamics involved in the connectivity of cells with the extracellular matrix is made possible through various support systems present in the cells viz., a) adherens junctions - responsible for cell-to-cell contact stability, transitory interconnections with neighbouring cells by forming a cadherin-catenin complex, comprised of cadherin, β-catenin, α-catenin and δ-catenin, which in conjunction form actin bundle. Another association through afadin and nectin has also been observed [11]. The effector key proteins involved in the dynamism as membrane and cytosolic bound tyrosine kinases phosphorylate β-catenin at nascent junctions. At the same time, phosphatases remove added phosphates from β-catenin and δ-catenin, as well as at Rho GTPases regulating the energy to activate catenins. b) On the contrary, tight junctions made of two transmembrane proteins-claudin and occludin maintain a fluidity barrier exchange over the movement of various transmembrane proteins between the cell surfaces (apical and basolateral) but are permeable to adjacent exchange of substances for maintaining polarity of the cell. Tight junction proteins 1 or ZO-1 interact with other proteins-F11, GJA3, GJA8, KIRREL, MLLT4, and TJP3. (c) Integrins build focal adhesions through FAK (focal adhesion kinase) in the cell with extracellular matrix (as composed of eighteen (α) alpha/eight (β) beta heterodimeric receptors) assisted through activation of auto-phosphorylation at Tyr397, binding position for the Src family kinases in PI3K and PLCγ. Through this combination of alpha and beta subunits, 24 types of integrins can be originated [12]. The deterioration of the adherens junctions that conserve epithelial cell proximity results in EMT is the loss of epithelial integrity by the proteolytic dissolution of matrix metalloproteinases (MMPs), which is modulated by EMT mediated TGF-β signaling. Additionally, the transcriptional end of genes encoding epithelial-specific proteins- E-cadherin, occludins and desmoplakins by pro-EMT transcription components such as SNAIL. Throughout EMT, epithelial adherens junctions are restored by N-cadherin that furnishes considerable junctional adaptability with increased cell motility. The activity of EMT necessitates builiding of the actin cytoskeleton through communication between the three ezrin, radixin and moesin (ERM) proteins together with CD44, a cell surface glycoprotein extansively present in cancer stem cells, and firmly balanced with cell motility and cancer metastasis, act as a receptor for extracellular proteoglycans such as hyaluronan and versican [13] (Fig. 1).
Epithelial-mesenchymal transition (EMT)
The known transcription factors such as TWIST, SLUG, SNAIL and ZEB have appeared to up-regulate the expression of various epithelial extracellular matrix (ECM) proteins, together with collagen 1, vitronectin and fibronectin and engage the proteases MMP-7, MMP-9 and MMP-14 to the surface where they degenerate ECM and basement membranes promoting tumor invasion and metastasis, with their action through cognate receptors like TGF-β, tyrosine kinase, etc., leading further to activation of intracellular pathways which through EMT cause upregulation of selected zinc finger, e.g., SNAIL1, SLUG, ZEB1, ZEB2) or basic helix-loop-helix e.g., TWIST1 transcription factors [14]. TGF-β/BMP signaling was initiated by forming heterotetrameric ligand-receptor complexes via the networking of ligands (e.g., TGF-β1, TGF-β2, BMPs) through Type I and Type II kinase receptors. These hetero-tetramers phosphorylate receptor guided SMADs (R-SMADs), together with co-SMAD (SMAD4), translocate the SMAD complex to the nucleus effecting the expression of target genes. SMADs (SMAD6/7), directly block the R-SMAD phosphorylation, thereby reducing the induction of EMT and its inhibition by the SMURF proteins (SMURF1/2) representing SMAD7 to the plasma membrane permitting its dynamic function with R-SMADS for the receptor binding [15-17]. Also, the few integrins are found to be up-regulated during EMT, including α5β1, which binds fibronectin and the integrins α1β1 and α2β1, collaborate with collagen I and block the involvement of E-cadherin complex. Cell co-operation with the ECM must be modified by ECM-related proteins; for example, SPARC, a glycoprotein, assists the association of collagen and α2β1 to stimulate EMT by preventing the SLUG expression in melanoma [18, 19]. SERPINE1 (PAI-1), another epithelial ECM segment, restrains the vitronectin to the integrin αvβ3, and its expression level in different types of diseases. Tissue inhibitor matrix metalloproteinase 1 (TIMP1), associated with CD63, causes EMT like cell transformation through the β1 integrin signal. TGF-β signaling pathways initiate EMT, oncogenic receptor tyrosine kinase of PI3K and RAS/RAF pathways regulate the malignant growth-promoting EMT. The RAS/RAF pathway start transcriptional activation of key EMT, while the PI3K pathway suppresses GSK3β phosphorylation of β-catenin targeting EMT. The TGF-β receptor initiates p38 MAPK stimulating the EMT translation factor FOXC2 (Forkhead box protein C2) [20, 21]. EMT down-regulates E-cadherin, driving the loss of epithelium that characterizes EMT. Likewise, lowering the expression of E-cadherin initiates oncogenic Wnt downstream signaling by dispensing and translocating β-catenin, from the adherens junctions to the nucleus. Activated Wnt expression is a distinct component of colorectal cancer showing overexpression of β-catenin, while in inactivated Wnt, unbound cytoplasmic β-catenin undergoes proteasomal degradation by phosphorylation and ubiquitination by β-catenin complex whereas, Wnt Frizzled receptor, GSK3β suppresses β-catenin complex activity. It translocates it to the nucleus to displace the Groucho/HDAC complex to initiate TCF/LEF translation of EMT effectors (SNAIL and N-Cadherin). Wnt/β-catenin associated EMT has been shown in various tumors, like osteosarcoma, gastric and prostate cancers. However, activation of EMT effectors-LEF1 is engaged with the microRNAs (miRNAs) involved in the expansion of EMT. EMT is also stimulated by the activation of the non-canonical STAT and MEK/ERK pathways and found to be increased in metastatic colon, lung, breast and liver cancer through binding of Wnt5A and Wnt5B to Fzd2 (Frizzled2) receptor. Wnt5A/Frizzled signaling has also appeared to trigger EMT through other non-canonical Wnt signaling pathways incorporating JNK and PKC signals in pancreatic cancer and melanoma. In tumors with augmented intracellular calcium levels and non-canonical Wnt signaling, less EMT expression prevails. Activation of PKC and CaMKII blocks β-catenin and stops the activation of TWIST and ZEB1 effectors. Juxtracine signaling pathway includes the initiation of NOTCH receptors, which finally displace KDM5A transcriptional repressor from a translation factor complex inducing EMT [22, 23] (Fig. 1).
Actin and Microtubule
GPCRs (G protein-coupled receptors), integrins, RTKs (receptor tyrosine kinases), and the semaphorin 1a receptor PlexinA, profoundly affect the cell movement and multiplication. The connection between the cytoskeleton and extracellular network through which integrins function to activate FAK (focal adhesion kinase) and Src kinase bring about phosphorylation of paxillin and the Crk-related substrate p130 Cas. The intracellular activated reaction is executed through the Rho complex of GTPases (Rho, Rac, and Cdc42) with their activators, like downstream protein kinase effectors, guanine nucleotide exchange factors (GEFs), through direct recruitment of the GTPases to actin proteins. These proteins manage the association of actin cytoskeleton proteins like cofilin, Arp2/3 complex, Ena/VASP, profilin, gelsolin, and forminins [24]. Various pathways can give rise to specific actin structures whose organization is significant for coordinated cell movement and integrity. Movement is controlled by myosin-actin elements in which tropomyosins stabilize out F-actin by preventing dynamizing factors. Uncontrolled activation of cytoskeletal pathways changes extracellular reactions in various diseases. Microtubules, the dynamic polymers of α/β-tubulin heterodimers in which GTP hydrolyzes on the β-tubulin subunit, are required for the intracellular vesicle transport and separation of chromosomes during the process of mitosis. Most of the microtubules are nucleated and a dynamic combination of depolymerisation and polymerisation is managed by either tubulin dimers or gathered microtubules incorporating stathmin, which sequesters tubulin and enhances microtubule components by the collapsin response mediator protein-2 (CRMP2) building microtubule tubulin dimers. Different proteins with microtubules contain MAP1C (microtubule-associated protein 1C) that make tau, keeping up microtubules in active state via MAP1B (Microtubule-associated protein 1B). A significant signaling pathway that controls microtubule subunits includes GSK-3β, a kinase commonly dynamic under basal development conditions. Numerous microtubule proteins and even non-microtubule proteins, for example-Xenopus microtubule-associated protein 215 (XMAP215); assemble through tubulin dimer to encourage its binding with plus-end tracking proteins (+TIPs), followed with the end binding protein (EB1). In the adenomatous polyposis coli (APC), a few non-motile kinesins from the kinesin-13 family through the mitotic centromere associated kinesin (MCAK) increase by immobilizing the association between the proto-filaments. In tubulin, a few post-translational alterations such as acetylation, poly-glutamylation and poly-glycylation appear to alter with microtubules organization [25] (Fig. 2).
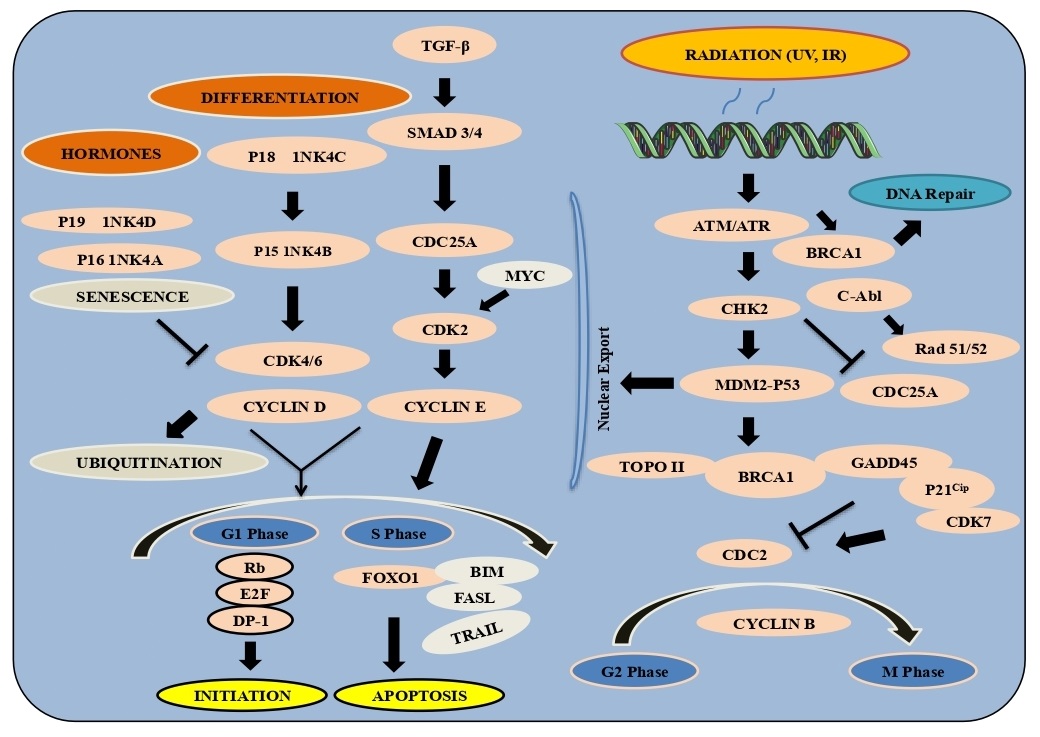
Cell cycle
Control of eukaryotic cell development and division includes circuits known as checkpoints that assure legitimate planning of the cell cycle. Entry through a checkpoint starting with one cell cycle stage then onto the next requires a planned arrangement of proteins that screen cell development and DNA fidelity. Uncontrolled cell division or damaged DNA can add to genomic heterogeneity and tumorigenesis. G1/S checkpoint controls the movement of cells through the restriction point into the DNA S-stage. During G1, the tumor silencer Rb restrains translation factor E2F. Phosphorylation of Rb by cyclin dependent kinases (CDK) in late G1 induce Rb separation and permit E2F-mediated translation to the S phase. The upstream signals, INK4 and Kip/Cip family inhibitors control CDK movement and suppress passage into the S phase. DNA damage starts reaction pathways through ATM/ATR (Ataxia-telangiectasia-mutated/ataxia telangiectasia and Rad3-related) and CHK1/2 kinases to prepare CDK movement to halt the cell cycle [26].
G2/M checkpoint arrest cells from entering mitosis (M) and initiates CDK1 (cdc2) bound to cyclin B enter into M-stage, whereas Wee1 and Myt1 kinases with cdc25 phosphatase block CDK1 and prevent entry into M-stage. DNA constricts different phosphorylate kinases Chk1/2 and the tumor suppressor protein p53 and restricts cdc25C. The checkpoint guarantees appropriate chromatid attachment before moving from metaphase to anaphase. SCF and APC/C protein with APC-cdc20 start anaphase by ubiquitin substrates, including cyclin B and securin. Also, the kinase Aurora A and the co-factor Bora initiate PLK1 (polo-like kinase 1) as cells approach the M-stage, stimulating the phosphatase cdc25, and downstream cdc2 action which drives the cell into mitosis. Significantly, DNA-PK/ATM/ATR kinases eventually inactivate the Cyclin B-cdc2 complex. Primarily, stopping the movement into mitosis is the checkpoint kinases which phosphorylate and inactivate cdc25, blocks the passing of cdc2, as well as phosphorylation of p53 and its separation from MDM2 and MDM4, passing DNA transcriptional movement. The transcriptional level of p53 is furthermore extended through acetylation by the co-activator complex p300/PCAF. The p53 downstream binds to the phosphorylated Cyclin B-cdc2 complex and GADD45 (growth arrest and DNA damage-inducible 45), separating the Cyclin B-cdc2 complex; and p21 Cip1, an inhibitor of the cyclin-dependent kinases including cdc2. In human cancer, mutated p53 demonstrate that this checkpoint is an essential parameter to tumor position, familial transformations in the DNA proteins, for example- the BRCA-family, ATM and fanconi anemia proteins further act as a key tumor suppressor checkpoint. G1/S cell cycle checkpoint controls the eukaryotic cells to progress through the G1 to go into the S phase. Two cell cycle kinase building blocks, CDK4/6-Cyclin D and CDK2-Cyclin E work in a complex that contains the retinoblastoma protein (Rb) and E2F. In G1-stage cells, hypo-phosphorylated Rb bind to the E2F-DP1 translation factors to form an inhibitory complex with HDAC to suppress key downstream transitions. Commitment to enter the S-stage happens through consecutive phosphorylation of Rb by Cyclin D-CDK4/6 and Cyclin E-CDK2 that separates the HDAC-repressor complex permitting translations required for DNA replication. Within the development factors, Akt can phosphorylate FoxO1/3 and restrict their activity by allowing stability to the cell movement. A large number of various checkpoint controls reveal the translation elements to establish expression from the INK4 or Kip/Cip groups of cyclin kinase inhibitors (CKIs). Remarkably, the oncogenic polycomb protein Bmi1 is a negative controller of INK4A/B in microorganisms and human cancer growth. Notwithstanding CKIs, TGF-β additionally restricts cdc25A translation, a phosphatase required for CDK regulation. At a primary assembly point with the DNA checkpoint, cdc25A is ubiquitinated by the SCF ubiquitin ligase complex downstream of the ATM/ATR/Chk-pathway. In mitosis, the M phase of the APC ubiquitin ligase elimination initiates GSK-3β to phosphorylate Cyclin D, which cause its ubiquitination and proteasomal degradation. Significantly, the assembly of Cyclin D1/CKD4/6 is investigated as a promising target for treatment as this checkpoint is invariantly deregulated in the human tumors [27, 28] (Fig. 3).
Kinases
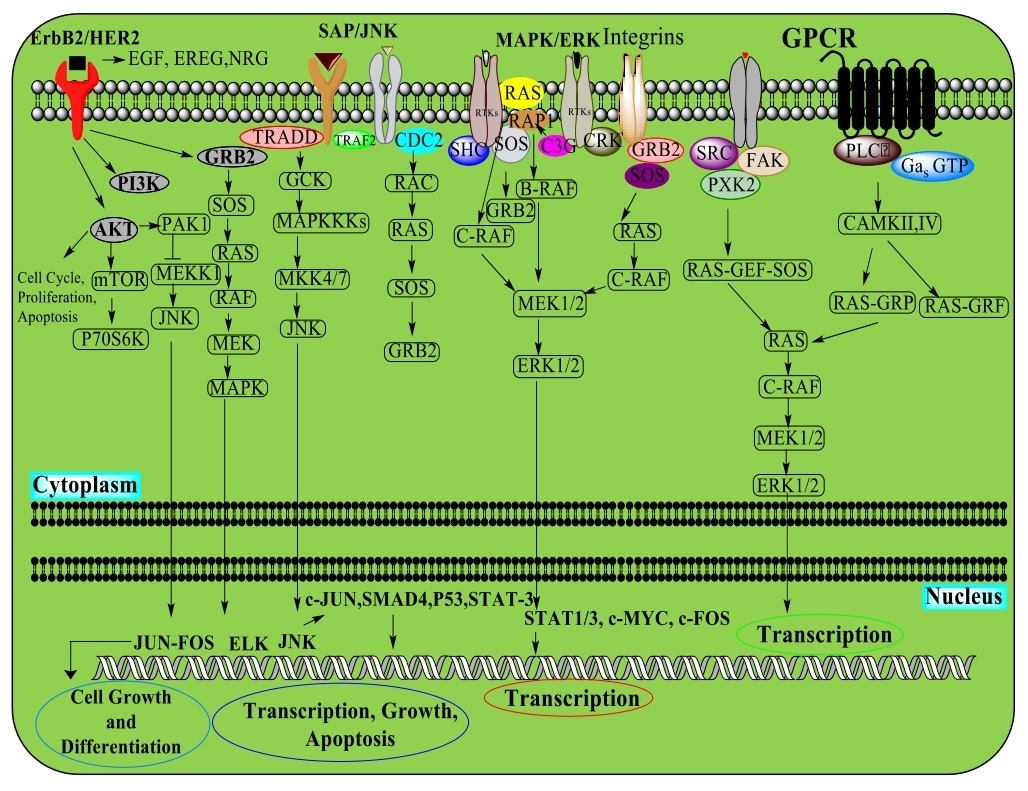
All Tyrosine kinases, working as interior effectors or cell surface receptors, direct protein function via exchange of ATP phosphate with the hydroxyl of tyrosine. They are single-pass, trans-membrane proteins that manage extracellular polypeptide ligands, cytoplasmic effectors and connector proteins. Ligand restriction promotes the receptor dimerization and auto-phosphorylation, resulting in a conformational change that ensues phosphorylation restriction for downstream effector proteins. EGFR (Epidermal growth factor receptor) is a well-determined tyrosine kinase under the ErbB receptor family. ErbB proteins bind with the extracellular ligands through homodimers or heterodimers with other family members. EGFR and ErbB4 attach with various ligands, while ErbB2 has no recognized ligand. ErbB3 has a functional kinase space and requires dimerization with an alternate ErbB for initiation. Different connector and effector proteins bind various targets inside the carboxy-terminal tail of a functioning kinase. MAPK/Erk signaling follows the GRB2 connector protein to EGFR at phospho-Tyr1068 and Shc protein and phosphorylates the EGFR at Tyr1148 and Tyr1173. The connector protein c-Cbl with the EGFR at Tyr1045 helps EGFR receptor ubiquitination and degradation. Non-receptor tyrosine kinases incorporate proteins from the Src family kinases, c-Abl and Jak kinases. The family kinases-Tec, bruton’s tyrosine kinase (BTK), E. coli tyrosine kinase (ETK), interleukin-2 inducible T-cell kinase (ITK), and tyrosine protein kinase (TXK), have pleckstrin homology area at the amino-terminal that recruits the kinase to the plasma surface, are essential for B cell and T cell receptor signaling. The other kinases FAK, FER, and activated CDC42 kinase 1 (ACK1) directs the cell development and movement, while non-receptor tyrosine kinase-1 (TNK-1) acts in pathways that adversely control cell development. Zeta-chain-associated protein kinase 70 (Zap-70) and Spleen tyrosine kinase (SYK) are associated with the various cellular reactions, while FES (Feline sarcoma oncogene) in hematopoiesis.
Human epidermal growth factor receptor (ErbB /HER)
ErbB1/EGFR/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4 are the four cell surface receptors of the ErbB receptor tyrosine kinase family. ErbB receptors move in the cell following ligand-receptor dimerization. Ligands-EGF, TGF-α, AR shows receptor specificity; neuregulins 1 to 4 links ErbB3 and ErbB4, while EGFR and ErbB4 are activated by the HB-EGF, epiregulin, and β-cellulin. Signaling through AKT, MAPK and numerous different pathways, ErbB receptors regulate cell proliferation, movement, separation, apoptosis and cell motility. ErbB was transformed in multiple types of cancer, making them significant targets. On the cell surface, ErbB family proteins, for instance, EGFR is moved into the nucleus where it works as a tyrosine kinase to phosphorylate and release PCNA, whereas bound ErbB2 communicates with importin β1 and Nup358 moves through endocytic vesicles. ErbB2, within the nucleus, alter the execution of different downstream conditions, including COX-2. Simultaneously, NRG or TPA activation promote ErbB4 cleavage by γ-secretase, which initiates apoptosis upon translocation to the nucleus. ErbB4, upon division, complexes with TAB2 (TGF-beta-activated kinase 1) and N-CoR (Nuclear Receptor Corepressor 1) to overcome the activation. ErbB receptors can be exchanged through receptor ubiquitination, dephosphorylation or movement of receptors from the cell surface by endosomal and lysosomal arrangements [29, 30].
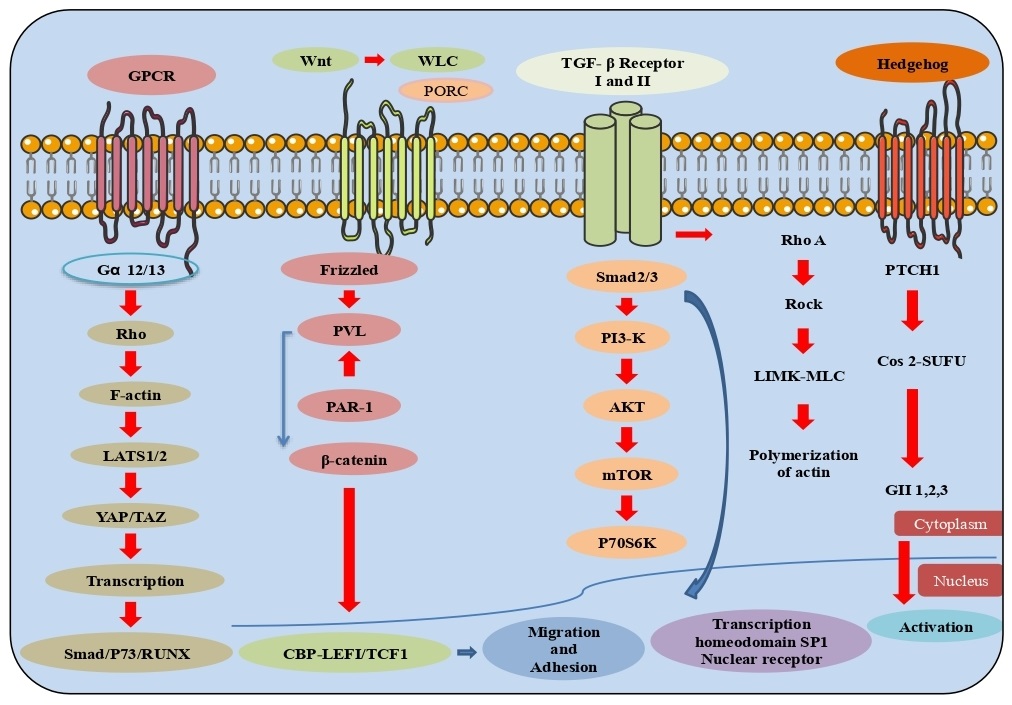
Stress-activated protein kinases (SAPK)/Jun amino-terminal kinases (JNK)
Stress activated protein kinases (SAPK)/c-Jun amino-terminal kinases (JNK) are individuals from the MAPK family, activated by various inflammatory cytokines and GPCR agonists. Stress signals are conveyed by GTPases of the Rho family- Rac, Rho, cdc42. Different MAPKs, MAPKKK, MEKK1 to 4, or MLK phosphorylates and activates MKK4 or MKK7, SAPK/JNK kinases. MKK4/7 can be generated from the germinal center kinase (GCK) family in GTPases. SAPK/JNK translocates to the nucleus, where it can direct the movement of numerous translation factors [31-33] (Fig. 4).
Mitogen-Activated Protein Kinase/extracellular-signal-regulated kinase (MAPK/ERK)
The MAPK/Erk pathway typically incorporates- Shc, GRB2 (Growth factor receptor-bound protein 2), Crk (CT10 Regulator of Kinase) connecting the receptor to a guanine nucleotide exchange factor- SOS, C3G transducing the GTP-restricting proteins Ras, Rap1, thus activate the MAPKKK (Raf), a MAPKK (MEK1/2), and MAPK (Erk) directing to the cytosol and translocates to the nucleus while phosphorylating various translation factors [34, 35] (Fig. 4).
G-Protein-Coupled Receptors (GPCR)
The G protein, upon activation exchanges GDP for GTP causing the separation of the GTP-bound α and β/γ subunits and activating the downstream signaling. Receptors coupled to various heterotrimeric G protein subtypes can cause various actions to initiate the G protein/MAPK, utilizing three unique classes of Tyr kinases. GPCRs use PLCβ to control the activation of PKC and CaMKII, which can have either stimulatory or inhibitory effects towards downstream of the MAPK pathway [36] (Fig. 4).
P38 Mitogen-Activated Protein Kinase (p38 MAPK)
p38 MAPKs α, β, γ, and δ are substituents from the MAPK family that are activated by cytokines. Within MAPK, ASK1 can control MKK3/6 that MAPKKK activates then phosphorylates and activates MKK3/6. ATF-2, STAT1, Max/Myc complex, MEF-2, and Elk-1 bind to p38 MAPK along with the HSP27, MAPKAPK-2 (MK2) and MAPKAPK-3 (MK3) [37-39] (Fig. 4).
5’ adenosine monophosphate-activated protein kinase (AMPK)
5’-AMP activated protein kinase (AMPK) is a heterotrimeric complex made of α, β and γ subunits. The γ subunit allosterically change substrate for phosphorylation on Thr172 of the α subunit by its major upstream AMPK kinase-LKB1. AMPK can be phosphorylated on Thr172 by CAMKK2 which impose changes in intracellular calcium by metabolic hormones including adiponectin and leptin. In low ATP levels, AMPK control pathways include unsaturated fat oxidation and autophagy. Contrarily, AMPK controls ATP biosynthetic processes including gluconeogenesis, lipid and protein synthesis. AMPK through direct phosphorylation controls the transcription by phosphorylating various co-activators and co-repressors acting as the primary regulator of lipid and glucose metabolism with potency to treat various cancers. It has been snared as an essential modulator through its association with mTOR and sirtuins [40, 41].
Mammalian target of rapamycin (mTOR)
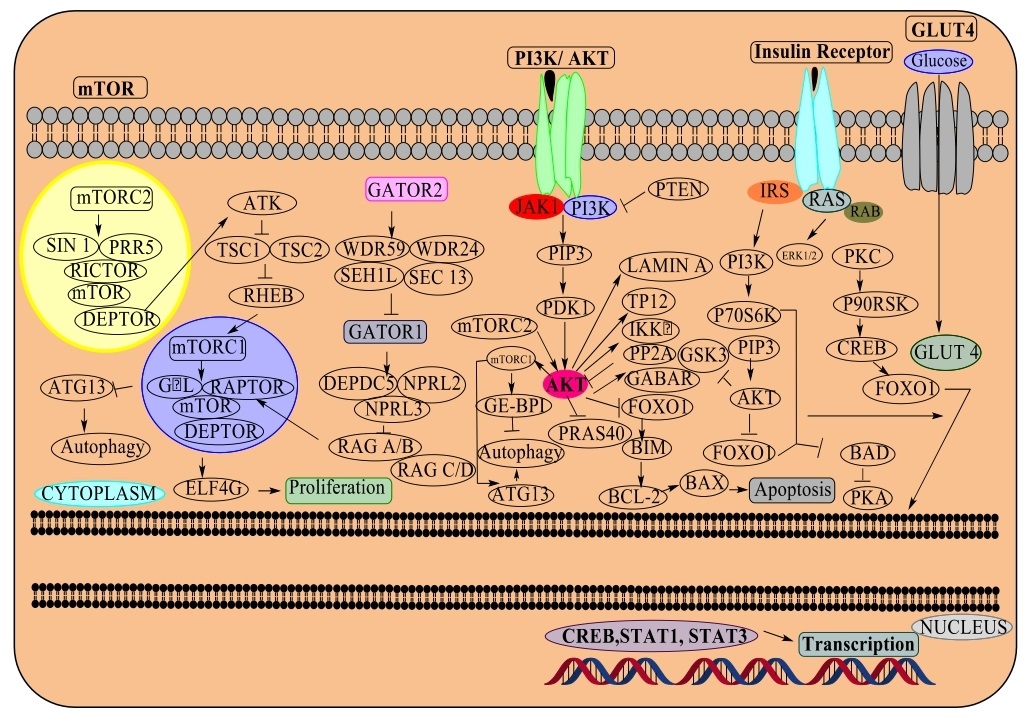
This pathway incorporates signals involved in the development factors, cell stress, protein synthesis and couples to the cell development by phosphorylation of substrates that lead to anabolic/catabolic processes corresponding to protein synthesis, lipid synthesis and autophagy. The serine/threonine kinase exists in two forms. The mTOR complex 1 (mTORC1) is made of mTOR, Raptor, GβL and DEPTOR and is blocked by rapamycin. The GTPase Rheb (Ras homolog enriched in brain) in its GTP-bound state triggers mTORC1 kinase action by GAP activity via TSC-1/TSC-2 (Tuberous sclerosis heterodimer). The mTOR complex 2, consisting of mTOR, GβL, Rictor, Sin1, PRR5/Protor-1 and DEPTOR, passes cell by directing Akt, cytoskeletal machinery by PKCα and controls transport by SGK1 phosphorylation [42, 43] (Fig. 5).
Phosphoinositide 3-kinase (PI3K)
Phosphatidylinositol is a lipid having an inositol ring, two unsaturated fat chains associated through a glycerol moiety, and a cytoplasmic structure present on the cell surface. Phosphatidylinositol is phosphorylated by a group of lipid kinases at the 3,4,5-hydroxyl of the inositol ring, with an assembly of phosphatidylinositol monophosphates (PI3P, PI4P and PI5P), diphosphates- PI (3,4) P2, PI (3,5) P2, PI (4,5) P2, and a triphosphate PI (3,4,5) P3 that are known as phosphoinositides. Phosphatidylinositol monophosphates are confined to the intracellular area by diffusion of endocytic vesicles and golgi complex, while di- and tri-phosphates located at the plasma membrane. PTEN and PI3K class I are receptor tyrosine kinase control AKT and are frequently changed in numerous types of cancer, cytoskeletal changes, actin remodelling, clathrin-mediated endocytosis, autophagy, cell division/cytokinesis and cell movement. Since its underlying mechanism as a proto-oncogene, the serine/threonine kinase PI3K-AKT (also known as protein kinase B or PKB) performs a significant role in managing metabolism, development, proliferation, translation and protein synthesis. The PI3K-AKT is initiated by RTKs, integrins, B and T cell receptors, various cytokine receptors, GPCRs that initiate the formation of phosphatidylinositol (3,4,5) trisphosphates (PIP3) by phosphoinositide 3-kinase (PI3K), serving as binding sites for pleckstrin-homology (PH) domain possessing proteins, including AKT and PDK1. PDK1 phosphorylates AKT at Thr308, causing movement of AKT at Ser473 by mTORC2. Components from the PI3K-related kinase (PIKK) family including DNA-dependent protein kinase (DNA-PK) can phosphorylate AKT at Ser473 and dephosphorylate by protein phosphatase 2A (PP2A) and the PH-area leucine-rich-continue containing protein phosphatases (PHLPP1/2). Similarly, the tumor suppressor phosphatase and tensin homolog (PTEN) prevent AKT activity by dephosphorylating PIP3. Dysregulation of the PI3K/AKT pathway is expressed in various human disorders, including cancer, diabetes, cardiovascular and neurological diseases. AKT1, AKT2 and AKT3 (isoforms of AKT) phosphorylate substrates containing RxRxxS/T phosphorylation site. As all AKT isoforms can phosphorylate PRAS40-proline-rich AKT substrate of 40kDa; the actin associated protein palladin and vimentin controls NF-κB by phosphorylating Tpl2 and IKKα. AKT directs cell development through its influence on the TSC-1/TSC-2 complex and mTORC. Also, AKT promotes cell growth by phosphorylation of CDK inhibitors (p21 and p27). It significantly inhibits apoptotic proteins like Bad or apoptotic signals produced by FoxO. Also, AKT has been appeared to control proteins associated with neuronal activity including the GABA receptor, ataxin-1 and huntingtin proteins [44, 45] (Fig. 5).
Protein Kinase C Signaling (PKC)
This essential protein structure incorporates an N-terminal associated with a C-terminal kinase. PKC contains an auto-inhibitory pseudo-substrate to regulate kinase action. The PKC is divided into three general families- cPKC- isoforms PKCα, PKCβ, and PKCγ have C1 and C2 spaces; cPKC requires diacylglycerol (DAG), a phospholipid to the C1 area and calcium to the C2 region. Novel PKC-nPKC-isoforms PKCδ, PKCε, PKCη and PKCθ additionally require DAG for initiation however, contain a novel C2 space. Exceptionally aPKC; isoforms PKCζ and PKCι/λ with a non-functional C1 and a C2 area. PKC is controlled through three phosphorylation at Thr500, Thr641 through auto-phosphorylation and at the C-terminal hydrophobic site Ser660 [46] (Fig. 5).
Insulin mediated signaling
For glucose and lipid digestion, insulin activates insulin receptor tyrosine kinase (IR), which selects and phosphorylates insulin substrate receptor (IRS) group of proteins - IRS 1, 2 and 4. PI3K has a significant role in insulin signaling mediated through the Akt/PKB and PKCζ. AKT trigger glycogen through the restriction of GSK-3 by mTOR downstream components, apoptotic mediators: Bad, FoxO, translation factors, GSK-3, and MST1, whereas AKT phosphorylates and prevent FoxO factor to control autophagy. Conversely, AMPK is known to control FoxO3 and initiate action of transcription. Insulin has development effect but is mostly intervened by the AKT through regulation of the RAS/MAPK pathway. The insulin pathway suppresses autophagy by the ULK1 kinase controlled by AKT and mTORC1 and stimulated by AMPK. Insulin triggers glucose assimilation in muscle and adipocytes by translocating GLUT4 vesicles to the plasma membrane. GLUT4 translocation includes the PI3K/AKT pathway and insulin receptor mediated phosphorylation of catabolite gene activator protein (CAP) and CAP:CBL:CRKII complex [47]. Also, insulin stops gluconeogenesis in the liver through the interference of CREB/CBP/mTORC2 generating unsaturated fatty acids and cholesterol through SREBP, USF1 and LXR. A negative feedback signals from Akt/PKB, PKCζ, p70 S6K, and the MAPK brings about serine phosphorylation and inactivation of the insulin substrate receptor. Regulation of eIF4E and p70 S6 Kinase in the mTORC1-S6K has a specific role in the control of translation by 5’ cap recognition by eIF4F, a trimeric protein complex made of eIF4E which link the 5ʹ cap, eIF4A and eIF4G proteins move the mRNA to eIF3 and bring out the circularization of mRNA with polyA binding protein (PABP). eIF4F is overcome by eIF4E translation initiation binding proteins (4EBPs), which in turn hypo-phosphorylates sequestering eIF4E preventing its binding with eIF4G. Furthermore, mTORC1 phosphorylates 4EBPs and permit its arrangement with the eIF4F with the start of translation. Also, mTORC1 adjacent to PDK1 phosphorylates S6 kinase, eIF4B, eIF4B- an activator of the eIF4A helicase, PDCD4 - an eIF4A inhibitor that is inhibited by phosphorylation; and SKAR, an mRNA splicing factor. Besides the mTORC1 pathway, the RAS-MAPK pathway is another significant controller of translation and is likely for the phosphorylation of eIF4B same as eIF4E by MNK kinases [48] (Fig. 5).
Janus kinases/Signal transducer and activator of transcription proteins
The cytokine receptors directing pathways emerge for the IL-6 or gp130 group of receptors, and co-regulate B cell separation, plasmacytogenesis. Receptor dimerization control of the JAKs phosphorylating receptor initiated by the inhibition of cytokines. The phosphorylated segment on the receptor acts as a docking terminus for the SH2 domain containing STAT-3 and for SH2-containing proteins that interface the receptor to MAP kinase, PI3K/AKT and other controlling pathways. Phosphorylated STATs dimerize and translocate into the nucleus to direct translation. Further, STAT-3 and 5 are constitutively initiated by tyrosine kinases in various tumors. On the other side, the JAK/STAT pathway mediates the cytokines similar to erythropoietin, thrombopoietin, and G-CSF which are protein targeted drugs and also utilized by the interferons acting as anti-viral and anti-proliferatives. IL-6 expression enhances the pathogenesis of the immune system to cause prostate cancer and myeloma. Crosstalk between cytokines and EGFR counterpart is found in some cancer cells and it has showed in glioblastoma cells overexpressing EGFR rescued from EGFR kinase inhibitors which are activated by JAK-2 to EGFR by FERM (4.1 protein, ezrin, radixin and moesin). JAK transformations are majorly involved in human cancers and found to be transformed in the JAK-2 pseudokinase domain that is V617F related to receptors for erythropoietin, thrombopoietin and G-CSF controlling erythroid, megakaryocytic and granulocytic differentiation that usually happens in polycythemia vera, thrombocythemia and idiopathic myelofibrosis. JAK-1 is found in T-cell lymphoblastic leukaemia, with a mutation in JAK1, 2, and 3 have additionally been recognized in acute lymphoblastic leukaemia (ALL) and down syndrome [49, 50] (Fig. 5).
Cell Death
Apoptosis is the programmed way in which cell progresses and brings about unusual events leading to cell death. This programmed process can be initiated through the death receptors like Fas, TNFαR, DR3, DR4, and DR5 by their receptor ligands through signaling by receptor oligomerization, whereas FasL conducts Fas trimerization activate initiator caspase-8 through the Fas-associated protein with death domain (FADD/MORT1) in contrast TNF-α and DR3 against apoptotic signals via TRADD/FADD and caspase-8. Also, TNF-α with its receptor may stimulate the NF-κB pathway by NF-κB-inducing kinase/ IκB kinase (NIK/IKK) complex. Caspases target proteins as a proteolytic apoptotic signal triggering initiation through cleavage and re-joining the p20 and p10 of an amino-terminal pro-domain. Caspases-2, 8, 9, 10 and 12 are initiator caspases that are coupled to upstream apoptotic signals and act as downstream effector or executioner caspases-3, 6, and 7. PARP and Lamin A and C are severed as markers of apoptosis. Caspases can be controlled by the inhibitors of apoptotic proteins (c-IAP1/2, livin, survivin) for ubiquitin-mediated action. Whereas inhibitor of apoptotic protein inhibitors-Smac/Diablo, reduce the expression of caspases and interfere to induce apoptosis. Control of mitochondrial cytochrome c includes permeabilization by Bcl-2 family proteins. Bcl-2 proteins further incorporate BH domain proteins (Bax, Bak) or a BH3 family (Bad, Bik, Bid, Puma, Bim, Bmf, and Noxa). Pro-survival Bcl-2 proteins incorporate Bcl-2, Bcl-xL, Bcl-2, and Bax may affect the voltage-dependent anion channel-VDAC, which controls cytochrome c releasing Bcl-w. Bax and Bak oligomerize at the mitochondria to permit cytochrome c into the cytosol bind to Apaf-1 and an initiation complex with caspase-9. BH3 family proteins are primary controllers of apoptosis and help cells by inhibiting apoptotic proteins including Bax and Bak into the mitochondria. The apoptotic Bcl-2 proteins Bad, Bid, Bax and Bim present in the cytosol translocate to mitochondria and signal the entry of cytochrome c. Following DNA damage, p53 starts off the transcription of bax, noxa and puma. The extrinsic pathway is stimulated through the death receptors including Fas, TNFαR, DR3, DR4 and DR5 by their different ligands through receptor oligomerization. FasL initiates caspase-8 by the protein FADD, in turn, activates caspase-8 and caspase-3, or it can separate Bid-an apoptotic Bcl-2 family protein, translocating it to mitochondria. DNA damage influences the outflow of P53 induced death domain containing protein 1 (PIDD) together with death domain-containing protein (CRADD), and caspase-2 stimulates the initiation of caspase-2. X-linked inhibitor of apoptosis protein (XIAP) restrains caspase-3, 7 and 9 through Smac/Diablo binds to XIAP supressing caspases. Caspase-11 causes the caspase-1 activation facilitating inflammatory reaction and undergoing apoptosis by caspase-3. Caspase-12 and 7 are signalled under ER stress conditions to functionalised AKT and p90RSK. AKT block Bad by direct phosphorylation preventing Bim by phosphorylating and the forkhead box of translation factors (FoxO) by upregulating apoptotic components FasL and Bim to undergo apoptosis. In mitochondria, the PI3K, JAKs and Src, Erk1/2 and PKC activate p90RSK pathway preventing the apoptotic Bcl-2 families Bad, Bax, caspase-9, GSK-3 and FoxO1.
On the other side, Necrotic cell death is an unprogrammed death having no involvement of caspases. RIPK1, RIPK3 and MLKL proteins are significantly engaged with necroptosis. Ubiquitination of RIPK1 by inhibitors of apoptosis proteins (IAP) cause initiation of NF-κB, and phosphorylation of RIPK1 at Ser320. Phosphorylation of RIPK3 at Ser227 is required for the activation of MLKL, an effector protein downstream of RIPK1 and RIPK3. RIPK1, RIPK3 by the TIR-domain-containing adapter-inducing interferon-β (TRIF), starts necroptosis by DNA-dependent activator of IFN regulatory factors/Z-DNA-binding protein 1 (DAI/ZBP1), which are sensors of viral RNA. From a disease perspective, neurodegeneration has been observed with necrostatin-1 (inhibitor of RIP1 kinase) in Alzheimer’s and Parkinson’s syndrome [51, 52].
During pyroptosis, extracellular components are expelled out from the cytoplasm via pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) to toll-like receptors (TLRs). There are four known inflammasome complex: cytosolic pattern recognition receptors (PRRs), a nucleotide binding domain (NBD) and leucine rich repeat (LLR) or AIM2-like receptor (ALR), an adaptor protein-apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1. After inflammasome formation, caspase-1 is proteolytically cleaved by the cytokines IL-1β and IL-18. In intracellular cytosolic lipopolysaccharide (LPS) gives rise to non-canonical pyroptosis causing cleavage of gasdermin D (GSDM-D) by caspase 4, 5 and 11 at the C-terminal domain by initiation of IL-1β [53].
Autophagy
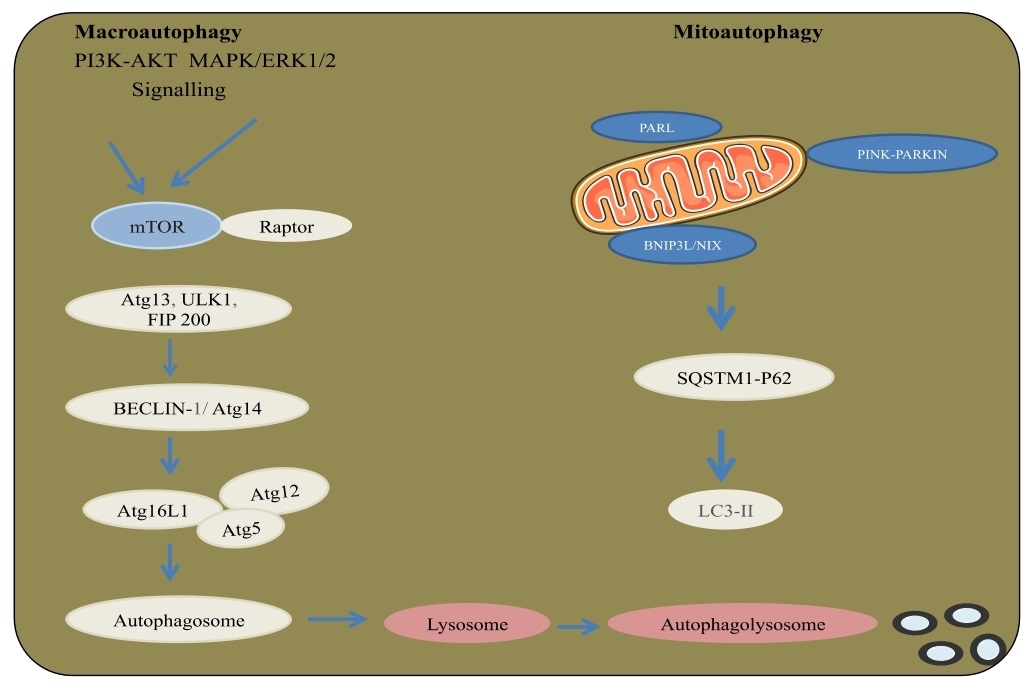
A catabolic process results in autophagosomes lysosomal breakdown in the cytoplasm. Autophagy has been related to physiological conditions like neurodegenerative disorders and cancer. The kinase mTOR with AKT and MAPK signaling is an essential regulator of autophagy with three related serine/threonine kinases, UNC-51-like kinase (ULK1, ULK2, UKL3) that acts downstream of the mTOR complex. ULK1 and ULK2 complex with the mammalian homolog of an autophagy-related gene-ATG and the protein focal adhesion kinase family interacting protein of 200 kDa (FIP200), an ortholog of yeast ATG17. Class III PI3K complex, containing Vps34, beclin-1(a mammalian homolog of yeast ATG6), p150- (a mammalian homolog of yeast Vps15), and ATG14 related protein or UV radiation resistance-associated gene protein (UVRAG), is required for the autophagy. The formation of autophagosome occurs through ATG12, ATG5 and LC3-II. ATG12 is conjugated to ATG5 in an ubiquitin-like response that requires ATG7 and ATG10 with E1 and E2 systems. Then, ATG12-ATG5 conjugate connects non-covalently with the ATG16 complex. LC3/ATG8 at its C-end by ATG4 protease form the cytosolic LC3-I. LC3-I is conjugated to phosphatidylethanolamine (PE), additionally in a ubiquitin-like response that requires ATG7 and ATG3. The lipid type of microtubule-associated protein 1A/1B-light chain 3 (LC3) known as LC3-II is joined to the autophagosome layer. Moreover, mitophagy is an autophagic process intended to clear damaged mitochondria from a cell. Upon damage, the protein PTEN-induced kinase 1 (PINK-1) was degraded through the activity of presenilins associated rhomboid-like protein (PARL) is stablized and regulate the E3 ligase parkin to start mitophagy. Polyubiquitination of mitochondrial membrane proteins by Parkinson protein 2, E3 ubiquitin protein ligase (Parkin) brings about the action of autophagy connector proteins SQSTM1/p62, NDP52, NBR1, and activating molecule in BECN1-regulated autophagy protein 1 (Ambra1) that bind to LC3 by their LC3 interacting region (LIR). Moreover, BNIP3 and BNIP3L/NIX contain LIRs allowing to select the autophagic network by a ubiquitin free component [54-56] (Fig. 6).
Senescence
A perpetual condition of halt in the cell cycle, senescence gives a resistance to keep tissue homeostasis and tumor development by permitting tissue alterations through the sequestration and removal of damaged constituents of cells. Human cells show replicative senescence, which is a limited proliferative condition brought about by the dynamic shortening of the telomeric ends triggering a DNA damage reaction (DDR) and arresting cell cycle through execution of reactions via chemotherapeutics, mitochondrial membrane potential (MMP) and reactive oxygen species (ROS), involving upregulation of signals through various intracellular pathways including the DDR components. ATR, ATM and p53 unite on the initiation of cyclin-dependent kinase inhibitors (CDKIs)-p16, p21, and p27 leading to hyperphosphorlyation of the retinoblastoma protein (RB). Albeit senescent cells do not multiply anymore, they remain metabolically dynamic and show remarkable morphological and physiological changes related to the cells state. Specifically, senescent cells have an expanded, flattened shape through the diminished expression of Lamin B1 protein. The aggregation of β-galactosidase, a consequence of changed lysosomal activity, is a sign of cell senescence. The development of senescence-associated heterochromatin foci (SAHF) directed through chromatin rearrangement act as a possible biomarker of cells experiencing oncogene promoted senescence and it can be identified by immunoreactivity for MacroH2A, Lysine 9 di/tri-methylated histone H3 (H3K9Me2/3) and heterochromatin protein 1 (HP1). DNA damage markers including the phosphorylation at Ser-139, and building histone variation γH2AX provide an assessment of senescence. Senescent cells frequently show remarkable changes in their secretome, leading to the senescence associated secretory phenotype (SASP). SASP involves the upregulation of cytokines (for example, IL-6/IL-1β), proteases (MMP3) and various development factors that through autocrine and paracrine networking changes the tissue microenvironment. It has been seen that SASP has been coupled to angiogenesis and extracellular matrix (ECM) to advance tumor cell movement [57-59].
Immune Checkpoint
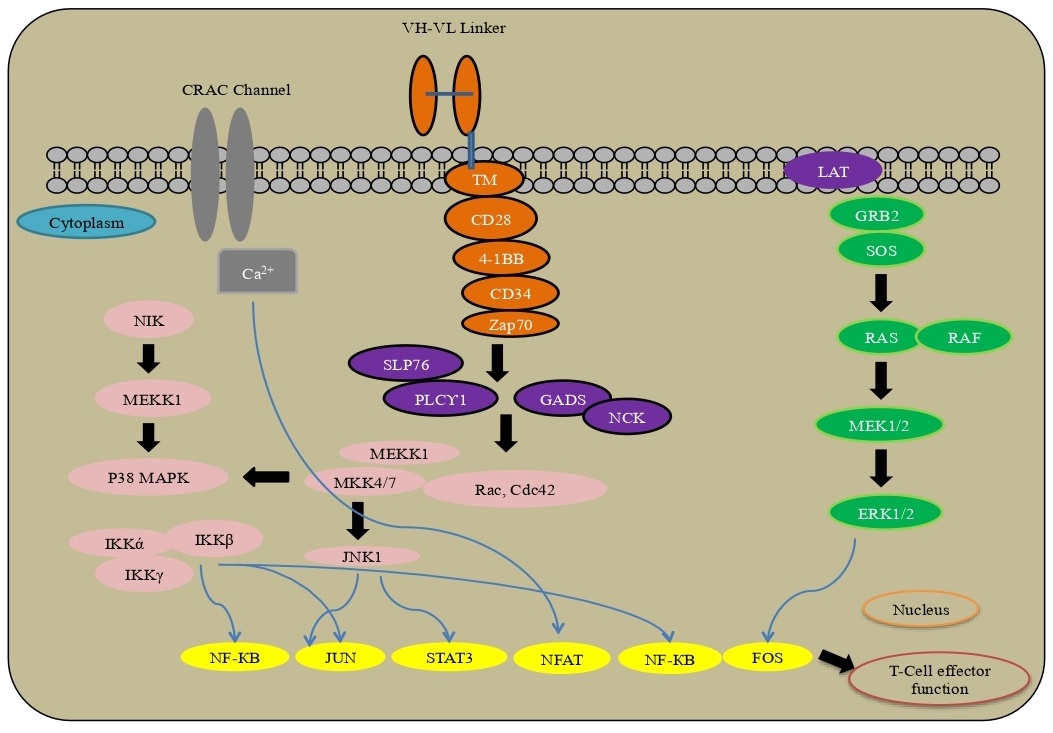
The tumor microenvironment has an intrinsic control under physiological conditions on immune cells, like CD8+ cytotoxic T lymphocytes and CD4+ adjuvant T cells, which associate through the T cell receptors (TCRs) with peptide antigens bound to major histocompatibility complex (MHC) on the exterior of antigen-presenting cells (APCs). T cells present a range of receptors like CD28, 4-1BB, ICOS, GITR, OX40 and APCs-CD80, CD86, OX40L, GITRL, 4-1BBL ICOSLG for T cell initiation. Alternately, the initiation process can be suppressed by co-inhibitory receptors- PD-1, TIM-3, CTLA-4 and LAG3. The interplay between PD-L1 or PD-L2 and the PD-1 receptor brings about down-regulation of TCR through dephosphorylation, whereas CTLA-4, a co-inhibitory receptor, competes with CD28 (a co-stimulatory receptor) to integrate with CD80 and CD86 ligands present on APCs. Upon ligand binding, CTLA-4 prevents T cell, while CD28 is required to activate the T cell. Inside the tumor microenvironment, malignant cells have inhibitory ligands and their receptors control T cell effector function to surpass resistance. Pharmacological modulators targeting the immune checkpoint, especially as monoclonal antibodies against PD-1 and CTLA-4, have been immensely explored as novel immunotherapy to treat cancer [60, 61]. CAR signaling, as promising immunotherapy, uses cells to treat the disease through the ex-vivo presentation and activation of a chimeric antigen receptor (CAR), the patient’s T cells are designed to focus on a particular surface antigen present on malignant cells.
A transmembrane space from CD8 or CD28 engages the CAR to the T-cell and associates the ARD to the intracellular compartment of the receptor. The receptor comprises an initiation area and one or different co-stimulatory spaces that provide ligand to control the change in T-cell transcriptional programs by carrying out a variety of downstream signaling. The site from the T-cell receptor CD3ζ chain is a typical element of the intracellular segments of CARs to drive T-cell cytotoxic capacities. A fundamental post-translational change upon ligand restricting is the phosphorylation of CD3ζ via zeta-chain associated protein kinase 70 (Zap-70) to downstream connector proteins. In co-stimulatory transcription, regulated through the PI3K/AKT, TNF receptor-related factor 2 (TRAF2), p38MAPK and JNK pathways including NF-κB, NFAT, STAT3, JUN and FOS to drive changes with the T-cell [62-64] (Fig. 7).
Pre-synaptic signaling
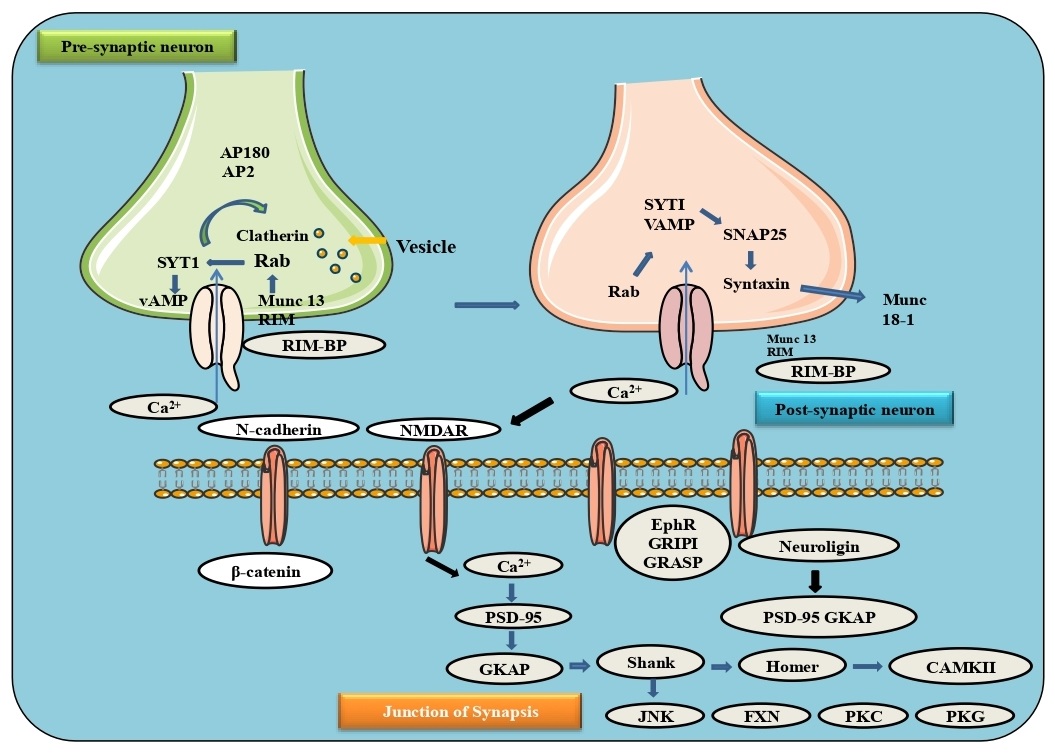
The neurotransmitter contains a presynaptic zone, calcium channels and the postsynaptic neuron with the ability to assemble and integrate synaptic signals. Intracellular vesicles containing synapses rapidly signal to the presynaptic layer and release their substance into the synaptic activity for neurotransmission. The binding of these vesicles is accomplished by the SNARE family and other chaperone proteins situated on both the vesicle and presynaptic cell layer. Synaptic vesicles docked in the dynamic zone by collaborating vesicle-related Rab3 (or Rab27) with RIM, which can bind to calcium channels by RIM-BP. The vesicle SNARE protein, VAMP-synaptobrevin, binds to SNARE proteins on the syntaxin 1 and SNAP25. The co-chaperone protein complexin (synaphin) and the calcium restricting protein synaptotagmin1 (SYT1) partner with SNARE proteins to bind the lipid membrane. An action potential in the presynaptic neuron opens voltage-gated calcium channels, where calcium binds to SYT1 and permits SYT1 to associate with the SNARE and the other proteins RIM, RIM-BP, and Munc13, forming physical association between the vesicle and calcium channels. Neurotransmitters can be recycled through transporters like excitatory amino acid transporters (EAATs) with reuptake of glutamate or a monoamine transporter, for example, serotonin reuptake transporter (SERT) for reuptake of serotonin or dopamine transporter (DAT) for reuptake of dopamine and again into the cytoplasm of the neuron [65-67] (Fig. 8).
Post-Synaptic Signaling
Excitatory synapses
In the postsynaptic layer, N-cadherins are trans-synaptically associated with Ephrin, Neurexin and Neuroligins 1 and 3. Glutamate is released from presynaptic vesicles in synaptic terminals as an excitatory neurotransmitter. There are three primary excitatory postsynaptic receptors in its synaptic terminals: α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA), N-methyl-D-aspartate (NMDA) and metabotropic glutamate receptors (mGluR). The AMPA receptors are ionotropic tetrameric glutamatergic receptors, and the PDZ (post synaptic density protein PSD95) space-containing AMPAR tetramers allow subunits to associate with the specific proteins and establish connections between the receptor and the cytoskeleton. For instance, two subunits, GluA2 and GluA3, associated with GRIP1, a connector protein with 7 PDZ, and PICK1. Through transmembrane AMPA receptor regulatory proteins (TARP) known as stargazin, AMPARs communicate with PSD-95, a key post-synaptic protein. Additional binding sites for GRIP1 include EphR and GRASP, a Ras guanine nucleotide exchange factor. Also, neuronal pentraxins (NP1, NARP, and NPR) can be delivered pre-synaptically and activate groups of AMPARs. Phosphorylation of AMPARs is another mechanism of AMPAR control, in addition to GRASP and neuronal pentraxins. In addition, CaMKII, JNK, FYN, PKC and PKG can all phosphorylate AMPARs, facilitating AMPAR recycling and TARP translocation from vesicles to synaptic terminals. In the postsynaptic neuron, AMPARs mediate depolarization and remove inhibitory cations from the NMDA pore, which allows the movement of Na+, Ca2+, and K+ into and out of the cell. Ca2+ outflow and CaMKII activation are the main factors contributing to long-term potentiation (LTP). Similar to AMPARs, NMDARs utilize kinases and phosphatases to carry their receptors to postsynaptic terminals. In addition to the glutamate receptor, NMDARs can bind postsynaptic density protein 95 (PSD-95) in the post-synaptic area. Synaptic GAP-Ras-GTPase is another protein that binds to the PDZ space of NMDAR-bound PSD-95. Metabotropic glutamate receptors (mGluRs) are G protein-coupled receptors that transmit signals through the cooperation of intracellular G proteins after binding glutamate in their extracellular N-terminal domain. The mGluRs contain eight subtypes, and the dimers they form with their C-terminal tails promote Ca2+ ion balance through inositol trisphosphate receptor (IP3R), an intracellular neuronal protein that connects mGluRs with IP3Rs. During post-synaptic excitatory processes, Ca2+ channels mainly through CamKII (calmodulin-dependent protein kinase II), which leads to downstream gene expression. CamKII phosphorylates key kinases necessary for synaptic plasticity and crosslinks F-actin fibers, as well as phosphorylating neuroligin 1, extending its surface to trigger new neurotransmitter production. Additionally, Ca2+ is taken up by IP3Rs in the endoplasmic reticulum, which is further activated by CamKII and AMPAR, leading to synaptic plasticity [68-71] (Fig. 8).
Inhibitory synapses
Post-synaptic GABA receptors (GABARs) and GLYRs are the main inhibitory post-synaptic receptors. They are both ligand-gated channels. It has heteropentamers, four transmembrane spaces, an N-terminal extracellular section, and an intracellular section between the third and fourth transmembrane helices. Synaptic connections to GABA or Glycine are found in the N-terminal space extracellularly. CAM molecules (transmembrane synaptic cell adhesion molecules; CAMs) cross over the inhibitory postsynaptic surface to the presynaptic membrane using Neurexin, Neuroligins 2, 3, and 4 a class of transmembrane synaptic cell adhesion molecules (CAMs). In addition to Neuroligin 2, Neuroligins 3/4 bind specifically to specific proteins in the postsynaptic structure to support further postsynaptic connectivity. During development, Neuroligin 2 cooperates with another transmembrane CAM, Slitrk3 (SLIT and NTRK like family member 3), and regulates inhibitory neural connections by collaborating with the axonal receptor protein tyrosine phosphatase, PTPδ. Neuroligin 2 binds GABARs, GLYRs, and Gephyrin within the inhibition postsynaptic space. GABARs, GLYRs, and tubulin bind multimeric Gephyrin, and collybistin, the GDP/GTP exchange factor, and proceed through Cdc42 related F-actin. Moreover, gephyrin is also complexed with the profilin and Mena, thereby increasing cytoskeletal association inside inhibitory postsynaptic density. Significantly, GABAR action itself promotes palmitoylation of gephyrin by DHHC-12, which results in the assembly of gephyrin and an increase in the inhibitory synaptic transmission. Inhibitory neurotransmission is established by the constant feedback loop between gephyrin association, GABAR (Gamma-aminobutyric acid receptor subunit alpha-1) and GABAR. Additional to Gephyrin, Neuroligin 2 binds MDGA1 (a glycosylphosphatidylinositol anchor with a MAM domain), a cellular molecule that provides steric hindrance to neurexins during the interaction between neuroligin and neurexins. Expression of MDGA1 represses inhibitory neurotransmitter development and serves as a synaptogenesis checkpoint. The excitatory postsynaptic receptor of GABARs is fundamental for the regulation and has the capacity for inhibitory neural associations. The intracellular receptor of GABARs is intervened by GABARAP (Gamma-aminobutyric acid receptor-associated protein precursor), a protein that collaborates with its intracellular space and increase in the GABARAP expression endocytosis, and lysosomal recycling. Ankyrin G inhibits GABAR endocytosis through GABARAP, increase the flow of GABARs and promotes GABAergic neurotransmitters. Significantly, GABAR is regulated by Ca2+ through NMDA receptors. This interplay between excitatory and inhibitory postsynaptic signals further emphasizes the significance of intracellular Ca2+ levels in excitation and inhibition. The serine/threonine kinase AKT manages different hormones and cytokines by regulating their related receptor tyrosine kinase (RTK), cytokine receptor, or GPCR and the lipid kinase PI3K, which produces PIP3 at the plasma membrane. AKT binds PIP3 through its pleckstrin homology (PH), bringing about the translocation of AKT to the surface. PDK1, through its PH space phosphorylates Akt via activation at Thr308. Second phosphorylation at Ser473 at the carboxy end is required for regulation and is done by the mTOR-rictor complex, mTORC2. PTEN, a lipid phosphatase that catalyzes the dephosphorylation of PIP3, is a significant negative controller of the AKT pathway. AKT action is contrarily controlled by the phosphatases PP2A and PHLPP, just as by the synthetic modulators wortmannin and LY294002, the inhibitors of PI3K [72, 73] (Fig. 8).
Angiogenesis
The development of new blood vessels from veins initiates chemotaxis of endothelial cells towards the tumorigenesis. Inside hypoxic conditions in the tumor, the dimeric protein complex of hypoxia inducible factor-1 (HIF-1) activates the angiogenic proteins incorporating vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) which advance vascular penetrability and endothelial cell development. Other factors like platelet derived growth factor (PDGF) and angiopoietin-1/2 (ANG-1/2) stimulate cell, while ephrin signaling assists newly formed veins by controlling motility and cell-cell attachment. Another function of HIF-1 is metalloproteinases (MMPs) networking that separates the extracellular matrix (ECM), and cause endothelial cell movement and releases development factors including specific integrins- αvβ3 found on the outside of angiogenic endothelial cells helping extracellular matrix to relocate and maintain. For example, various angiogenic factors, VEGF and MMPs released into the microenvironment initiate tumor associated macrophages (TAMs) to facilitate angiogenesis. Pericytes work on the basolateral surface of ECs, managing vasoconstriction and extension angiogenesis of the tumor under physiologic conditions. A ligand for PDGF receptors situated on the pericyte surface causes VEGF signals through the endothelial VEGF receptor. Neutrophils involve cell invasion and advance tumor angiogenesis through numerous components via incorporating of MMPs in the tumor microenvironment, releasing VEGF and other angiogenic factors. Different resistant cell types involving B and T cells impact angiogenesis using VEGF-A, bFGF, MMP9, interferon γ (IFNγ), interleukin-17 (IL-17), adipocytes releasing cytokines, chemokines, and hormones [74-76].
Fibrosis
Fibrosis is the scarring of tissue brought about by the extracellular lattice (ECM) proteins by myofibroblasts and immune system responses. Fibrosis influences all tissues of the body and brings about organ disintegration. The key signaling pathways that control fibrogenesis has distinguished potential to stem the movement of fibrosis and help to re-establish the function of the cell. Myofibroblasts from various origin, including fibroblasts, mesenchymal cells, and fibrocytes assist the replacement of parenchymal cells. In the cell milieu, cytokines and other development factors are discharged with transforming growth factor-beta (TGF-β) family and Wingless/Int-1 (Wnt1), acting as the foremost effectors of the fibrotic process. TGF-β and Wnt1 bind to their related cell surface receptors and start downstream signaling prompting the translocation of Smad2/3 and CBP/β-Catenin transcriptional modulators. This results in the upregulated expression of ECM proteins including collagen, laminin, and fibronectin transduction through the cell surface integrin receptors passing the hippo signaling pathway and its downstream effectors. Yes-associated protein 1 (YAP1) and Tafazzin (TAZ), further enhance the upregulation of profibrotic including connective tissue growth factor (CTGF) and Platelet-derived growth factor (PDGF) progress initiation and multiplication of myofibroblast by PI3K/AKT/mTOR pathway. Instances of conditions related to neurotic fibrosis incorporate non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), idiopathic pulmonary fibrosis (IPF), alcoholic liver disease (ALD) and renal fibrosis. Besides, fibrosis has also been involved in tumor cell growth which through fibrotic extracellular matrix (ECM), stimulate cell division and alter the cells to promote tumor growth and development [77-79].
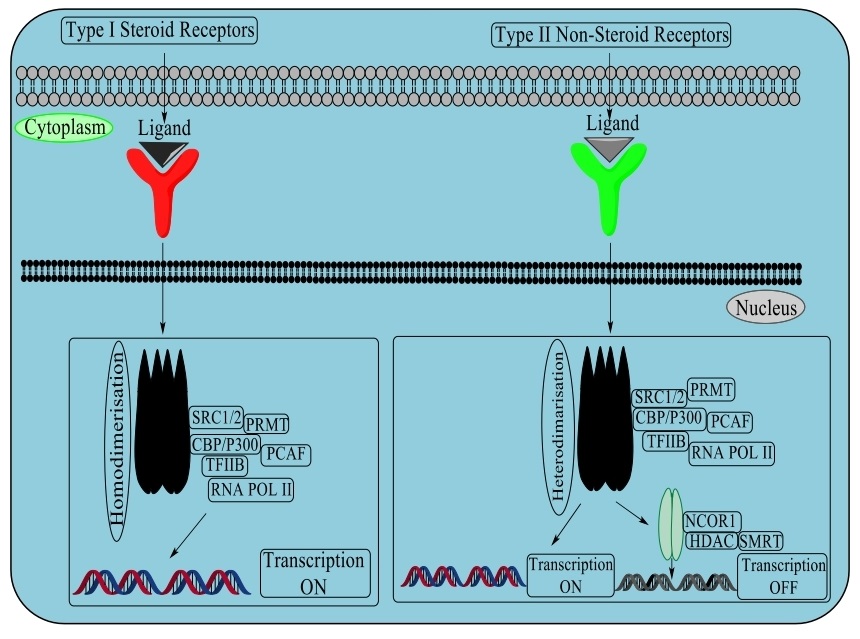
Type I and Type II nuclear receptors
This superfamily consists of ligand translation factors which work in cell separation and invasion related to various diseases. Individuals from this family contain an N-terminal transactivation space, an area of zinc-finger DNA and a C-terminal ligand. Type I or steroid receptors incorporate the estrogen receptor, androgen receptor, progesterone receptor, mineralocorticoid receptor and glucocorticoid receptor. The ligand receptor complex separates from the HSP90, where it homodimerizes and transactivates the co-activator, acetyltransferases. Type II or non-steroid receptors incorporate various hormones, for instance: the thyroid hormone receptors-TR α/β, retinoic acid receptors- RAR-α/β/γ, vitamin D receptor- VDR, and peroxisome proliferator activated receptors- PPAR-α/β/γ, which heterodimerize with the retinoid X receptor- RXR with histone deacetylases- HDACs and other co-repressors that target DNA through ligand restriction in disconnecting co-repressor, chromatin inactivation and initiation of transcriptional activity [80]. Various other nuclear receptors where the endogenous ligands have not been recognized but ought to control translation either as monomers or homodimers recruiting chromatin co-activators and assimilating heterodimer accomplice-SHP, Rev-Erb α/β, testicular receptor 2 and 4, homologue of the drosophila tailless gene (TLX), photoreceptor cell specific nuclear receptor (PNR), chicken ovalbumin upstream promoter transcription factor binding to the ovalbumin promoter (COUP-TF1/2), nuclear receptor subfamily 2, group F, member 1 (N2RF1), nuclear receptor related 1 protein (NURR1), neuron-derived orphan receptor 1 (NOR1), estrogen related receptor (ERR α, β, γ), and germ cell nuclear factor (GCNF). Protein kinase C (PKC) control various cell reactions and incorporates an amino-terminal associated with a carboxy-terminal kinase. PKC contain an auto-inhibitory pseudo-substrate that binds to restrict the kinase action. Three PKC isoforms PKC-α, PKC-β and PKC-γ, have functional C1 and C2 confirmatory areas requiring diacylglycerol (DAG) and a phospholipid to the C1 component while calcium ion (Ca2+) confine to the C2 region. Other discovered PKC catalyst isoforms nPKCs, PKC-δ, PKC-ε, PKC-η, and PKC-θ involve DAG for regulation but with no Ca2+ induction. Atypical protein kinase C (aPKC) with isoforms PKC-ζ and PKC-ι/λ contain non-functional C1 and C2 components. In DNA damage progression and oxidative stress, it has been seen that PKC-δ progress apoptosis through the initiation of the p53 mediated pathway and inhibition of the expression of pro-survival proteins- AKT, CDK-1 and Cyclin D1. In transmembrane signaling through phospholipase, phosphoinositide specific phospho-lipase C (PLC) hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) and generate the auxiliary inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG), which are controlled by phosphorylation. The four groups of PLCs have been recognized which are PLC-β, PLC-γ, PLC-δ and PLC-ε. Phosphorylation of PLC-β3 at Ser1105 by protein kinase PKA/C inhibits action through phosphorylation of PLC-γ at Tyr 771, 783 and 1245 by epidermal growth factor receptor (EGFR), non-receptor tyrosine kinases (nRTKs) spleen tyrosine kinase (SYK), janus kinase (JAK), feline sarcoma oncogene (FES), and activated CDC42 kinase 1 (ACK-1) bringing about initiation.
Moreover, the PLC-β subfamily activated by α, β, γ subunits of heterotrimeric G-proteins carries out a significant role in G-protein coupled receptor (GPCR) signaling. In G-protein coupled receptors, signaling transmission to MAPK/ERK requires G protein interchange of GDP for GTP to cause the separation of the GTP-bound α/β/γ subunits. Receptors coupled to various heterotrimeric G protein subtypes activate the G protein/MAPK utilizing three classes of tyrosine kinases. Proto-oncogene tyrosine-protein kinase (Src) family kinases are selected after initiation of PI3Kγ by β/γ subunits and by receptor cross-activation of receptor Tyr kinases, or an integrin including proline-rich tyrosine kinase-2 (Pyk2) as well as focal adhesion kinase (FAK). GPCRs can utilize PLC-β to control protein kinase-C (PKC) and calcium/calmodulin-dependent protein kinase II (CaMKII) through either stimulatory or inhibitory products for the downstream MAPK signaling pathway [81, 82] (Fig. 9).
RNA regulation and translational regulationThe events in translational control are the association between 5’ mRNA, the pre-initiation complex and the initiator tRNA to the start codon. These are managed by different eukaryotic initiator factors (eIF), effector kinases and inhibitors. Subunit proteins eIF4A, eIF4G and eIF4E, involve the eIF4F complex that binds the 5’ mRNA complex with other proteins and progresses into the pre-initiation complex. The eIF4F is supressed by development factors that control the movement of upstream kinase effectors, including AKT, PI3K, p70 S6 kinase, p90RSK and mTOR. The mammalian target of rapamycin (mTOR) kinase builds mTORC complex 1/2 pass eIF4F (E74 like ETS transcription factor 4) by activating upstream components that complex together and prevent proteins in promoting eIF4F growth. The mTORC1 complex incorporates mTOR kinase by the raptor and G protein beta subunit-like (Gβl), proline-rich Akt substrate of 40 kDa (PRAS40) and DEP domain-containing mTOR-interacting protein (DEPTOR), while the mTORC complex 2 contains rapamycin insensitive companion of mammalian target of rapamycin (RICTOR) and sin domain. It has been seen that ribosomal protein S6 kinase beta-1 (p70S6) stimulated through mTORC complex 1 alleviates programmed cell death 4 (PDCD4/H731) of eIF4A RNA helicase and initiate eIF4B to collaborate with both the eukaryotic initiation factor 3 (eIF3) protein complex and 45kDa initiation factor 4A (eIF4A) to start off eIF4A RNA helicase activity. Inactivation of the eukaryotic translation initiation factor-4E (eIF4E) inhibitor and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) by mTORC complex 1 causes eIF4E entry and regulating into eukaryotic initiation factor 4F (eIF4F). Through the AKT, mTORC2 stops the action of TSC2/TSC1, a heterodimer that supress mTOR action via GTPase Ras homolog enriched in brain (Rheb). The initiator tRNA binds to the ribosome toward the starting codon to frame the 43S pre-initiation complex, inhibited by eIF2. Trimeric eIF2 is comprised of tRNA/mRNA cooperating (β) and GTP/GDP heterotrimeric G-proteins-γ proteins. Phosphorylation of eukaryotic initiation factor 2 (eIF2α) by many upstream kinases-heme-regulated inhibitor (HRI), protein kinase R (PKR), PKR-like endoplasmic reticulum kinase, (PERK) and general control non-depressible 2 (GCN2) follows dsDNA and makes eIF2 inactive and prevents translation. The eIF2 is facilitated by eIF2B through the exchange of GDP for GTP promoting connectivity loop between the eIF2 complex and tRNA where eIF2B action is suppressed by GSK-3β phosphorylation via linking with eukaryotic translation initiation factor-5 (eIF5) as a GDP inhibitor [83].
A few post-transcriptional alterations are available in eukaryotic mRNA, where N
6-methyladenosine (m
6A) is a well-known mammalian RNA transcriptome, very predominant in mRNAs and some non-coding RNAs. It has been seen that close association to stop codons in 3’ untranslated regions of mRNA inside the mRNA exons communicates with the RNA motif, i.e., RRACH (R-purine, A-m
6A, and H-A/C/U). The N
6-methyladenosine (m
6A) alteration directs various phases of mRNA digestion and drives T-cell separation, undifferentiated cell dissociation, epithelial-mesenchymal progression, adipogenesis and neurogenesis. The expansion of N
6-methyladenosine (m
6A) to mRNA is a reversible remodelling directed by methyltransferases to introduce the epi-modifications and require erasers or demethylases to remove them. Incorporation of demethylases
via the complex-Methyltransferase complex is made of different subunits including methyltransferase-like 3 (METTL-3/14/16) and Wilms tumor 1-associated protein (WTAP). METTL14 act as a connector preventing the substrate and helps in methyltransferase movement. WTAP guides the complex to mRNA directing to the nucleus and promote mRNA splicing and RNA processing. The YTH space containing proteins that act as m
6A restricting proteins can be divided into the significant classes: YTHDC1/2 and YTHF proteins. YTHDC1 proteins are found in the nucleus, coordinating with mRNA join through YTHDC2 and YTHDF proteins which has cytoplasmic translational effect of m
6A altered mRNAs. Three paralogs DF1/2/3 from YTH domain-containing family protein from Drosophila (YTHDF) proteins has been reported. The eukaryotic initiation factor 3 (eIF3) assemble on the 40S smallest subunit either directly by m
6A inside 5′ untranslated region (5’ UTR) of mRNAs or through a YTHDF1. Several heterogeneous nuclear ribonucleoproteins A2/B1(HNRNPA2B1/HNRNPC/HNRNPG) and insulin like growth factor-2 mRNA binding proteins 1, 2 and 3 (IGF2BP1/2/3) found in the nucleus prevents m
6A proteins; causes interchange in mRNA permitting HNRNPC and HNRNPG or insulin like growth factor 2 (IGF2) restrict proteins to m
6A to affect mRNA separately. Besides, HNRNPA2B1 binds with small non-coding RNA molecule (miRNA) either directly to m6A or by non-methylated arrangement. As investigated, changes in the m
6A result in the tumorigenesis of various origins, including lung, myeloid leukaemia and glioblastoma [84].
For instance, ZNF217 cooperates with METTL3 in breast cancer cells and thus repress the m
6A transcripts, SOX2, c-MYC, KLF4 and NANOG. In lung cancer cells, small ubiquitin like modifier 1 (SUMO1) accelerate the post-translational transformations of METTL3 while supressing m
6A. Messenger RNA (mRNA) is directed by various RNA binding proteins (RBPs), starting with translation in which co-activators binds to DNA enhancer elements that signal RNA polymerase II to initiate RNA transcription complex. The strands of the DNA helix were separated incorporating a pre-mRNA in the DNA strand by joining RNA nucleotides in a 5’ to 3’ manner. During transcription, pre-mRNAs are assembled into ribonucleoprotein (RNP) incorporating 5’ mRNA capping and polyadenylation. After the formation of mRNP, it translocates through the nuclear pore to the cytoplasm guided by the cooperation of key RBPs including NXF1, XPO1, and PHAX. In the cytoplasm, RNPs associated with RBPs binds to the 5’ cap and 5’ untranslated complex. This multi-protein system encourages the association between mRNAs and the 40S ribosomal subunit that processes the transcript for the starting codon by enrolling the 60S ribosomal subunit and the amino acid chain initiation. The exon junction complex (EJC) found on RNPs comprises RBPs including CASC3, MAGOH, RBM8A, EIF4A3, and PYM1. The mRNAs in the cytoplasm are affected by the RNA interference (RNAi) system. In RNAi, non-coding RNAs are controlled by dicer which directs their enzymatic degradation by the RNA-induced silencing complex (RISC). Nonsense-mediated mRNA decay (NMD) activated by an EJC signal downstream of the end codon on an RNP during translation controlled by the length of the 3’ untranslated region of the mRNA to distinguish and takes out mRNA transcripts (containing stop codons), consequently preventing the transcription of shortened proteins [85, 86].
The eIF2 complex directs mRNA translation
via eukaryotic initiation factor 2 (eIF2-GTP) and methionyl-tRNA synthetase (Met-tRNAi) that construct the ternary complex (TC) with the 40S ribosomal subunit. The eukaryotic translation initiation factor 1 (eIF1, 1A, F5, F3) form the 43S pre-initiation complex (PIC) to assess the mRNA, an untranslated region (UTR) for an AUG start codon, then eIF2 hydrolyzes GTP to GDP through the help of GTPase initiating eukaryotic translation initiation factor-5 (eIF5) which subsequently separates from the mRNA and extends the polypeptide chain by 60S ribosomal subunit. For another initiation, eIF2B act on both GDP dissociation inhibitor (GDI) and guanine nucleotide exchange factor (GEF), allowing GDP for GTP exchange on eIF2 through phosphorylation of eIF2α by four kinases, namely a) protein kinase R (PKR) (dsRNA), b) protein kinase RNA-like endoplasmic reticulum kinase (PERK) (ER stress), c) general control non-derepressible 2 (GCN2) (amino corrosive starvation), and d) heme-regulated eIF2α kinase (HRI). The eukaryotic initiation factor 2 (eIF2α-GDP) restricts the accessibility of the ternary complex by the translation factors (activating transcription factor 4) ATF-4 and C/EBP homologous protein (CHOP). At the post-transcriptional level, nine key eukaryotic initiation factors (eIFs) and the ribosomal complex process through the development of the 48S complex from the 43S initiation complex and mRNA followed together with the 60S subunit allowing polypeptide chain arrangement. Increased levels of Ca
2+ or cyclic adenosine monophosphate (cAMP) can inhibit the activity of eukaryotic elongation factor 2 (eEF2) through the regulation of AMPK [87-89].
UbiquitinationCell proteins use the ubiquitin-proteasome system (UPS) to inspect a framework for the disposal of misfolded proteins directed by signaling pathways. This signaling assumes to be associated with the cell division, transcriptional processes and apoptosis. Ubiquitin is an exceptionally monitored 76 amino acid polypeptide that can be covalently connected to numerous cell proteins by the ubiquitination procedure. It is an ATP dependent and is completed by ubiquitin activating protein (E1)-thioester bond with ubiquitin-conjugating compound (E2) by the arrangement of an iso-peptide between the carboxy-end of ubiquitin and the ε-amino group of a lysine. The ubiquitin ligase (E3) remodels a subset of substrate proteins and passes to the 26S proteasome for degradation. Mono/poly-ubiquitination in which substrate proteins are connected to ubiquitin utilizing seven specific ubiquitin lysine residues (Lys 6, 11, 27, 29, 33, 48 and 63). In polyubiquitinated proteins, the ubiquitin is linked through lysine 48/63 (K48/63) polyubiquitin chains to target proteins for proteasomal degradation and protein-protein interactions. Deubiquitinase (DUBs) reverses ubiquitination by expelling ubiquitin from its substrate protein through five sub-families viz., universal stress protein (USP), ubiquitin carboxyl-terminal hydrolase (UCH), ovarian tumor protein (OTU) and metalloproteases group Jab1/Mov34/Mpr1Pad1N-terminal+ (MPN+) (JAMM) each with a particular tissue and substrate. The 26S proteasome is an ~2 MDa complex that fits in as the proteolytic arm of the ubiquitin-proteasome substructure. It comprises two sub-units- the 19S regulatory molecule (RP) and the 20S subunit core particle (CP). Small ubiquitin-like modifiers (SUMO-1/2/3) and neural precursor cell expressed NEDD8 can be covalently joined to proteins utilizing an E1, E2, E3 conjugation framework [90-92].
Future ProspectsIn the cancer moon-shot regime, various targeted therapies are coming along with the support of different drug companies but as we are growing, cancer is also advancing in establishing its resilience. Major challenges faced by companies in signal transduction are that the pathways are not entirely understood, and chief pathway players in various cancers have to be discovered as a potential drug targets. Bioinformatics has revolutionized the efforts to scan signal transduction pathways concerning to the disease. Besides, RNA integrated signaling involving genomics and proteomics has made efforts in mapping pathway(s) to decipher a variety of interacting proteins complexes in their native states and obtain sequence information encompassing evolution.
ConclusionEach cell represents an essential unit of life composed of multiple interactions and cross-talk cascades involved in the chemical reaction(s) resulting in signal transduction. However, when the pathway is disrupted, it influences the disease state whether chemically or physically. Various drugs and personalised medicines have been proposed to target these processes or pathways, but they have not entirely succeeded in targeting these receptors due to their undruggable nature. To develop effective therapeutics, targeting signals through artificial intelligence can be an innovative approach for developing a benchmark for signal transduction.
AbbreviationsAPC (Adenomatous polyposis coli); APC (Antigen-presenting cell); ATF-2 (Activating transcription factor 2); ATM (Ataxia telangiectasia mutated); ATP (Adenosine triphosphate); ATR (Ataxia telangiectasia and Rad3-related protein); BCL-2 (B-cell lymphoma 2); CAM (Cell adhesion molecules); CAP (Catabolite activator protein); CAR (Chimeric antigen receptors); CDK (Cyclin-dependent kinases); COX (Cyclooxygenase); CRADD (CASP2 and RIPK1 domain containing adaptor with death domain); CRMP2 (Collapsin response mediator protein); CTGF (Connective tissue growth factor); CTLA-4 (Cytotoxic T-lymphocyte-associated protein 4); DDR (DNA damage response); DEPTOR (DEP domain containing mTOR interacting protein); ECM (Extracellular matrix); EGFR (Epidermal growth factor receptor); EMT (Epithelial mesenchymal transition); EZH2 (Enhancer of zeste homolog 2); FADD (Fas-associated protein with death domain); FERM (F for 4.1 protein, E for ezrin, R for radixin and M for moesin); FES (Feline sarcoma oncogene); FOX (C2, Forkhead box protein C2); GABA (Gamma-Aminobutyric acid); GCK (Glucokinase); GCNF (Germ cell nuclear factor); GCSF (Granulocyte-colony stimulating factor); GDI (Guanine nucleotide dissociation inhibitors); GEF (Guanine nucleotide exchange factors); GLUT (Glucose transporter); GPCR (G protein-coupled receptor); GSDMD (Gasdermin D); HDAC (Histone deacetylases); HIF-1 (Hypoxia-inducible factor-1); HSP90 (Heat shock protein 90); IL (Interleukins); IRS1 (Insulin receptor substrate 1); ITK (Interleukin-2-inducible T-cell kinase); JAK (Janus kinases); JNK (c-Jun N-terminal kinase); KDM5A (Lysine Demethylase 5A); LAG3 (Lymphocyte-activation gene 3); LIR (LC3-interacting region); LTP (Long-term potentiation); MAP (Microtubule-associated proteins); MAPK (Mitogen-activated protein kinase); MCAK (Mitotic centromere–associated kinesin); MHC (Major histocompatibility complex); MMP (Matrix metalloproteinases); MST1 (Macrophage-stimulating 1); mTOR (Mammalian target of rapamycin); NBD (Nucleotide binding domain); NCOR1 (Nuclear receptor corepressor 1); NFAT (Nuclear factor of activated T-cells); NF-Κβ (Nuclear factor kappa-light-chain enhancer of activated B cells); NMDA (N-methyl-D-aspartate receptor); NMDAR (N-methyl-D-aspartate receptor); NRG (Neuregulin 1); NTRK (Neurotrophic tropomyosin receptor kinase); PABP (Poly (A)-binding protein); PAMP (Pathogen associated molecular patterns); PARL (Presenilins-associated rhomboid-like protein); PARP (Poly [ADP-ribose] polymerase 1); PCNA (Proliferating cell nuclear antigen); PD-1 (Programmed cell death protein 1); PDGF (Platelet-Derived Growth Factor); PDK1 (Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1); PI3K (Phosphoinositide 3-Kinase); PIDD (P53-induced protein with a death domain); PINK (PTEN-induced kinase 1); PIP3 (Phosphatidylinositol (3,4,5)-trisphosphate); PLK (Polo-like kinases); PNR (Photoreceptor cell-specific nuclear receptor); PTEN (Phosphatase and tensin homolog deleted on chromosome 10); RB1 (Retinoblastoma protein1); RHEB (Ras homolog enriched in brain); RHO (Ras homologous); RIPK3 (Receptor interacting serine/threonine kinase 3); ROS (Reactive oxygen species); RTK (Receptor tyrosine kinases); SAHF (Senescence-associated heterochromatin foci); SAPK (Stress and mitogen activated protein kinases); SASP (Senescence associated secretory phenotype); SERT (Serotonin transporter); SH2 (Src Homology 2); SNARE (Soluble N-ethylmaleimide-sensitive factor activating protein receptor); STAT (Signal transducer and activator of transcription); SYK (Spleen Associated tyrosine kinase); TAB2 (TGF-beta activated kinase 1); TARP (TCR gamma alternate reading frame protein); TCF/LEF (T-cell factor/lymphoid enhancer factor); TCR (T-cell receptor); TGF-β (Transforming growth factor beta 1); TJP3 (Tight junction protein 3); TLR (Toll-like receptors); TNF-α (Tumor necrosis factor); TRAF2 (TNF receptor-associated factor 2); TWIST1 (Twist-related protein 1); UPS (Ubiquitin/Proteasome System); UVRAG (UV radiation resistance-associated gene); VEGF (Vascular endothelial growth factor); WNT (Wingless-related integration site); XIAP (X-linked inhibitor of apoptosis protein); YAP (Yes-associated protein); ZAP (Zinc finger antiviral protein); ZEB (1, Zinc finger E-box-binding homeobox 1).
Acknowledgements
Author’s Contributions
AQ: Conceptualization, design and drafted; SMS: Reviewing and editing; SKS: Design and guidance. All authors read and approved the final manuscript.
Funding
This work was supported by the Indian Institute of Integrative Medicine, CSIR, Jammu-180001, and Academy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India.
Disclosure Statement
The authors declare that no conflicts of interest exist.
References
1 Sever R, Brugge JS: Signal transduction in cancer. Cold Spring Harb Perspect Med 2015;5:a006098.
https://doi.org/10.1101/cshperspect.a006098 |
| |
2 Macaluso M, Paggi M, Giordano A: Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncogene 2003;22:6472-6478.
https://doi.org/10.1038/sj.onc.1206955 |
| |
3 Lee WS, Yang H, Chon HJ: Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med 2020;52:1475-1485.
https://doi.org/10.1038/s12276-020-00500-y |
| |
4 Müller IE, Rubens JR, Jun T, Graham D, Xavier R, Lu TK: Gene networks that compensate for crosstalk with crosstalk. Nat Commun 2019;10:4028.
https://doi.org/10.1038/s41467-019-12021-y |
| |
5 Fritz V, Fajas L: Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 2010;29:4369-4377.
https://doi.org/10.1038/onc.2010.182 |
| |
6 Jack ID, Shaw AT: Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15:81-94.
https://doi.org/10.1038/nrclinonc.2017.166 |
| |
7 Haider T, Pandey V, Banjare N, Gupta PN, Soni V: Drug resistance in cancer: mechanisms and tackling strategies. Pharmacol Rep 2020;72:1125-1151.
https://doi.org/10.1007/s43440-020-00138-7 |
| |
8 Saputra EC, Huang L, Chen Y, Kellogg LT: Combination Therapy and the Evolution of Resistance: The Theoretical Merits of Synergism and Antagonism in Cancer. Cancer Res 2018;78:2419-2431.
https://doi.org/10.1158/0008-5472.CAN-17-1201 |
| |
9 Ochsner SA, Abraham D, Martin K, Ding W, McOwoti A, Kankanamge W, Wang Z, Andreano K, Hamilton RA, Chen Y, Hamilton A, Gantner ML, Dehart M, Qu S, Hilsenbeck SG, Becnel LB, Bridges D, Maayan A, Huss JM, Stossi F, et al.: The Signaling Pathways Project, an integrated 'omics knowledgebase for mammalian cellular signaling pathways. Sci Data 2019;6:252.
https://doi.org/10.1038/s41597-019-0193-4 |
| |
10 Qin S, Jiang J, Lu Y, Nice EC, Huang C, Zhang J, He W: Emerging role of tumor cell plasticity in modifying therapeutic response. Sig Transduct Target Ther 2020;5:228.
https://doi.org/10.1038/s41392-020-00313-5 |
| |
11 Lamouille S, Xu J, Derynck R: Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178-196.
https://doi.org/10.1038/nrm3758 |
| |
12 Gonzalez DM, Medici D: Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal 2014;7:re8.
https://doi.org/10.1126/scisignal.2005189 |
| |
13 Martin TA, Harrison G, Mansel RE, Jiang WG: The role of the CD44/ezrin complex in cancer metastasis. Crit Rev Oncol Hematol 2003;46:165-186.
https://doi.org/10.1016/S1040-8428(02)00172-5 |
| |
14 Fenouille N, Tichet M, Dufies M, Pottier A, Mogha A, Soo JK, Rocchi S, Mallavialle A, Galibert MD, Khammari A, Lacour JP, Ballotti R, Deckert M, Deckert ST: The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS One 2012;7:1-15.
https://doi.org/10.1371/journal.pone.0040378 |
| |
15 Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Machado LO, Yang J: Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011;19:372-386.
https://doi.org/10.1016/j.ccr.2011.01.036 |
| |
16 Khammanivong A, Gopalakrishnan R, Dickerson EB: SMURF1 silencing diminishes a CD44-high cancer stem cell-like population in head and neck squamous cell carcinoma. Mol Cancer 2014;13:260.
https://doi.org/10.1186/1476-4598-13-260 |
| |
17 Chandhoke AS, Karve K, Dadakhujaev S, Netherton S, Deng L, Bonni S: The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial-mesenchymal transition in a sumoylation-regulated manner. Cell Death Differ 2016;23:876-888.
https://doi.org/10.1038/cdd.2015.152 |
| |
18 Kurrey NK, Amit K, Bapat SA: Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol 2005;97:155-165.
https://doi.org/10.1016/j.ygyno.2004.12.043 |
| |
19 Noë V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M: Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci 2001;114:111-118.
https://doi.org/10.1242/jcs.114.1.111 |
| |
20 Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A: TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 2005;16:1987-2002.
https://doi.org/10.1091/mbc.e04-08-0658 |
| |
21 Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K: Two major Smad pathways in TGF-beta superfamily signaling. Genes Cells 2002;7:1191-1204.
https://doi.org/10.1046/j.1365-2443.2002.00599.x |
| |
22 Polakis P: The many ways of Wnt in cancer: Curr Opin Genet Dev 2007;17:45-51.
https://doi.org/10.1016/j.gde.2006.12.007 |
| |
23 Schmalhofer O, Brabletz S, Brabletz T: E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev 2009;28:151-166.
https://doi.org/10.1007/s10555-008-9179-y |
| |
24 Poukkula M, Kremneva E, Serlachius M, Lappalainen P: Actin-depolymerizing factor homology domain: a conserved fold performing diverse roles in cytoskeletal dynamics. Cytoskeleton (Hoboken) 2011;68:471-490.
https://doi.org/10.1002/cm.20530 |
| |
25 Rottner K, Stradal TE: Actin dynamics and turnover in cell motility. Curr Opin Cell Biol 2011;23:569-578.
https://doi.org/10.1016/j.ceb.2011.07.003 |
| |
26 Besson A, Dowdy SF, Roberts JM: CDK inhibitors: cell cycle regulators and beyond. Dev Cell 2008;14:159-169.
https://doi.org/10.1016/j.devcel.2008.01.013 |
| |
27 Reinhardt HC, Yaffe MB: Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009;21:245-255.
https://doi.org/10.1016/j.ceb.2009.01.018 |
| |
28 Malumbres M, Barbacid M: Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;3:153-166.
https://doi.org/10.1038/nrc2602 |
| |
29 Arteaga CL, Engelman JA: ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014;25:282-303.
https://doi.org/10.1016/j.ccr.2014.02.025 |
| |
30 Avraham R, Yarden Y: Feedback regulation of EGFR signaling: decision making by early and delayed loops. Nat Rev Mol Cell Biol 2011;12:104-117.
https://doi.org/10.1038/nrm3048 |
| |
31 Davies C, Tournier C: Exploring the function of the JNK (c-Jun N-terminal kinase) signaling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans 2012;40:85-89.
https://doi.org/10.1042/BST20110641 |
| |
32 Engström W, Ward A, Moorwood K: The role of scaffold proteins in JNK signaling. Cell Prolif 2010;43:56-66.
https://doi.org/10.1111/j.1365-2184.2009.00654.x |
| |
33 Haeusgen W, Herdegen T, Waetzig V: The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol 2011;90:536-544.
https://doi.org/10.1016/j.ejcb.2010.11.008 |
| |
34 McKay MM, Morrison DK: Integrating signals from RTKs to ERK/MAPK. Oncogene 2007;26:3113-3121.
https://doi.org/10.1038/sj.onc.1210394 |
| |
35 Keyse SM: Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev 2008;27:253-261.
https://doi.org/10.1007/s10555-008-9123-1 |
| |
36 Goldsmith ZG, Dhanasekaran DN: G protein regulation of MAPK networks. Oncogene 2007;26:3122-3142.
https://doi.org/10.1038/sj.onc.1210407 |
| |
37 Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA: p38 (MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 2009;15:369-379.
https://doi.org/10.1016/j.molmed.2009.06.005 |
| |
38 Cuadrado A, Nebreda AR: Mechanisms and functions of p38 MAPK signaling. Biochem J 2010;429:403-417.
https://doi.org/10.1042/BJ20100323 |
| |
39 Huang G, Shi LZ, Chi H: Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine 2009;48:161-169.
https://doi.org/10.1016/j.cyto.2009.08.002 |
| |
40 Cantó C, Auwerx J: AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 2010;67:3407-3423.
https://doi.org/10.1007/s00018-010-0454-z |
| |
41 Carling D, Mayer FV, Sanders MJ, Gamblin SJ: AMP-activated protein kinase: nature's energy sensor. Nat Chem Biol 2011;7:512-518.
https://doi.org/10.1038/nchembio.610 |
| |
42 Hoeffer CA, Klann E: mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 2010;33:67-75.
https://doi.org/10.1016/j.tins.2009.11.003 |
| |
43 Laplante M, Sabatini DM: Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 2013;126:1713-1719.
https://doi.org/10.1242/jcs.125773 |
| |
44 Luca AD, Maiello MR, D'Alessio A, Pergameno M, Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT signaling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets 2012;16:17-27.
https://doi.org/10.1517/14728222.2011.639361 |
| |
45 Mendoza MC, Er EE, Blenis J: The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 2011;36:320-328.
https://doi.org/10.1016/j.tibs.2011.03.006 |
| |
46 Dissanayake SK, Wade M, Johnson CE, O'Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, Jiang Y, Nickoloff BJ, Taub DD, Trent JM, Moon RT, Bittner M, Weeraratna AT: The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem 2007;282:17259-17271.
https://doi.org/10.1074/jbc.M700075200 |
| |
47 Rowland AF, Fazakerley DJ, James DE: Mapping insulin/GLUT4 circuitry. Traffic 2011;12:672-681.
https://doi.org/10.1111/j.1600-0854.2011.01178.x |
| |
48 Cheng Z, Tseng Y, White MF: Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 2010;21:589-598.
https://doi.org/10.1016/j.tem.2010.06.005 |
| |
49 Yu H, Pardoll D, Jove R: STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009;9:798-809.
https://doi.org/10.1038/nrc2734 |
| |
50 Sansone P, Bromberg J: Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol 2012;30:1005-1014.
https://doi.org/10.1200/JCO.2010.31.8907 |
| |
51 Kaufmann T, Strasser A, Jost PJ: Fas death receptor signaling: roles of Bid and XIAP. Cell Death Differ 2012;19:42-50.
https://doi.org/10.1038/cdd.2011.121 |
| |
52 Pradelli LA, Bénéteau M, Ricci JE: Mitochondrial control of caspase-dependent and independent cell death. Cell Mol Life Sci 2010;67:1589-1597.
https://doi.org/10.1007/s00018-010-0285-y |
| |
53 Kurokawa M, Kornbluth S: Caspases and kinases in a death grip. Cell 2009;138:838-854.
https://doi.org/10.1016/j.cell.2009.08.021 |
| |
54 Alers S, Löffler AS, Wesselborg S, Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012;32:2-11.
https://doi.org/10.1128/MCB.06159-11 |
| |
55 Ding WX, Yin XM: Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012;393:547-564.
https://doi.org/10.1515/hsz-2012-0119 |
| |
56 Feng D, Liu L, Zhu Y, Chen Q: Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res 2013;319:1697-1705.
https://doi.org/10.1016/j.yexcr.2013.03.034 |
| |
57 Campisi J: Aging, cellular senescence, and cancer. Annu Rev Physiol 2013;75:685-705.
https://doi.org/10.1146/annurev-physiol-030212-183653 |
| |
58 Faget DV, Ren Q, Stewart SA: Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019;19:439-453.
https://doi.org/10.1038/s41568-019-0156-2 |
| |
59 Coppé JP, Desprez PY, Krtolica A, Campisi J: The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99-118.
https://doi.org/10.1146/annurev-pathol-121808-102144 |
| |
60 Nirschl CJ, Drake CG: Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy: Clin Cancer Res 2013;19:4917-4924.
https://doi.org/10.1158/1078-0432.CCR-12-1972 |
| |
61 Francisco LM, Sage PT, Sharpe AH: The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010;236:219-242.
https://doi.org/10.1111/j.1600-065X.2010.00923.x |
| |
62 Srivastava S, Riddell SR: Engineering CAR-T cells: Design concepts. Trends Immunol 2015;36:494-502.
https://doi.org/10.1016/j.it.2015.06.004 |
| |
63 Benmebarek MR, Karches CH, Cadilha BL, Lesch S, Endres S, Kobold S: Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int J Mol Sci 2019;20:1283.
https://doi.org/10.3390/ijms20061283 |
| |
64 Ramello MC, Benzaïd I, Kuenzi BM, Moreno ML, Kandell WM, Santiago DN, Saldana MP, Darville L, Fang B, Rix U, Yoder S, Berglund A, Koomen JM, Haura EB, Daga DA: An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal 2019;12:eaap9777.
https://doi.org/10.1126/scisignal.aap9777 |
| |
65 Sudhof TC: The presynaptic active zone. Neuron 2012;75:11-25.
https://doi.org/10.1016/j.neuron.2012.06.012 |
| |
66 Sudhof TC: A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med 2013;19:1227-1231.
https://doi.org/10.1038/nm.3338 |
| |
67 Jahn R, Fasshauer D: Molecular machines governing exocytosis of synaptic vesicles. Nature 2012;490:201-207.
https://doi.org/10.1038/nature11320 |
| |
68 Hell JW: CaMKII: claiming center stage in postsynaptic function and organization. Neuron 2014;81:249-265.
https://doi.org/10.1016/j.neuron.2013.12.024 |
| |
69 Song I, Huganir RL: Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 2002;25:578-588.
https://doi.org/10.1016/S0166-2236(02)02270-1 |
| |
70 Won S, Incontro S, Nicoll RA, Roche KW: PSD-95 stabilizes NMDA receptors by inducing the degradation of STEP61. Proc Natl Acad Sci U S A 2016;113:E4736-E4744.
https://doi.org/10.1073/pnas.1609702113 |
| |
71 Huganir RL, Nicoll RA: AMPARs and synaptic plasticity: the last 25 years. Neuron 2013;80:704-717.
https://doi.org/10.1016/j.neuron.2013.10.025 |
| |
72 Li J, Han W, Pelkey KA, Duan J, Mao X, Wang YX, Craig MT, Dong L, Petralia S, McBain CJ, Lu W: Molecular Dissection of Neuroligin 2 and Slitrk3 Reveals an Essential Framework for GABAergic Synapse Development. Neuron 2017;96:808-826.
https://doi.org/10.1016/j.neuron.2017.10.003 |
| |
73 Tseng WC, Jenkins PM, Tanaka M, Mooney R, Bennett V: Giant ankyrin-G stabilizes somatodendritic GABAergic synapses through opposing endocytosis of GABAA receptors. Proc Natl Acad Sci U S A 2015;112:1214-1229.
https://doi.org/10.1073/pnas.1417989112 |
| |
74 Raza A, Franklin MJ, Dudek AZ: Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol 2010;85:593-598.
https://doi.org/10.1002/ajh.21745 |
| |
75 Sakurai T, Kudo M: Signaling pathways governing tumor angiogenesis. Oncology 2011;81: 24-29.
https://doi.org/10.1159/000333256 |
| |
76 Van Hinsbergh VW, Koolwijk P: Endothelial sprouting and angiogenesis: matrix metalloproteinases in the lead. Cardiovasc Res 2008;78:203-212.
https://doi.org/10.1093/cvr/cvm102 |
| |
77 Wynn TA, Ramalingam TR: Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 2012;18:1028-1040.
https://doi.org/10.1038/nm.2807 |
| |
78 Piersma B, Bank RA, Boersema M: Signaling in Fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front Med 2015;2:1-14.
https://doi.org/10.3389/fmed.2015.00059 |
| |
79 Schuster S, Cabrera D, Arrese M, Feldstein AE: Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349-364.
https://doi.org/10.1038/s41575-018-0009-6 |
| |
80 Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM: PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 2013;19:557-566.
https://doi.org/10.1038/nm.3159 |
| |
81 Goldsmith Z, Dhanasekaran D: G Protein regulation of MAPK networks. Oncogene 2007;26:3122-3142.
https://doi.org/10.1038/sj.onc.1210407 |
| |
82 Marinissen MJ, Gutkind JS: G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 2001;22:368-376.
https://doi.org/10.1016/S0165-6147(00)01678-3 |
| |
83 Dreyfuss G, Kim VN, Kataoka N: Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 2002;3:195-205.
https://doi.org/10.1038/nrm760 |
| |
74 Lukong KE, Chang KW, Khandjian EW, Richard S: RNA-binding proteins in human genetic disease. Trends Genet 2008;24:416-425.
https://doi.org/10.1016/j.tig.2008.05.004 |
| |
85 Lykke-andersen S, Jensen TH: Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 2015;16:665-677.
https://doi.org/10.1038/nrm4063 |
| |
86 Chang YF, Imam JS, Wilkinson MF: The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 2007;76:51-74.
https://doi.org/10.1146/annurev.biochem.76.050106.093909 |
| |
87 Steitz TA: A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol 2008;9:242-253.
https://doi.org/10.1038/nrm2352 |
| |
88 Wek RC, Jiang HY, Anthony TG: Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006;34:7-11.
https://doi.org/10.1042/BST0340007 |
| |
89 Sonenberg N, Hinnebusch AG: Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009;136:731-745.
https://doi.org/10.1016/j.cell.2009.01.042 |
| |
90 Amm I, Sommer T, Wolf DH: Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys Acta 2014;1843:182-196.
https://doi.org/10.1016/j.bbamcr.2013.06.031 |
| |
91 Budhidarmo R, Nakatani Y, Day CL: RINGs hold the key to ubiquitin transfer. Trends Biochem Sci 2012;37:58-65.
https://doi.org/10.1016/j.tibs.2011.11.001 |
| |
92 Burrows JF, Johnston JA: Regulation of cellular responses by deubiquitinating enzymes: an update. Front Biosci 2012;17:1184-200.
https://doi.org/10.2741/3980 |