×
![]()
Corresponding Author: Carlos E. Irarrázabal
Facultad de Medicina, Universidad de los Andes, Av. Mons. Álvaro del Portillo 12.455, Las Condes, Santiago (Chile)
Tel. +562 22618 1607 , E-Mail cirarrazabal@uandes.cl
1400W Prevents Renal Injury in the Renal Cortex But Not in the Medulla in a Murine Model of Ischemia and Reperfusion Injury
Consuelo Pastena,b Mauricio Lozanoa Gonzalo P. Méndezc Carlos E. Irarrázabala,b
aLaboratorio de Fisiología Integrativa y Molecular, Programa de Fisiología, Centro de Investigación e Innovación Biomédica, Universidad de los Andes, Santiago, Chile, bFacultad de Medicina, Universidad de los Andes, Santiago, Chile, cAnatomía Patológica, Laboratorio Inmunocel, Santiago, Chile
Introduction
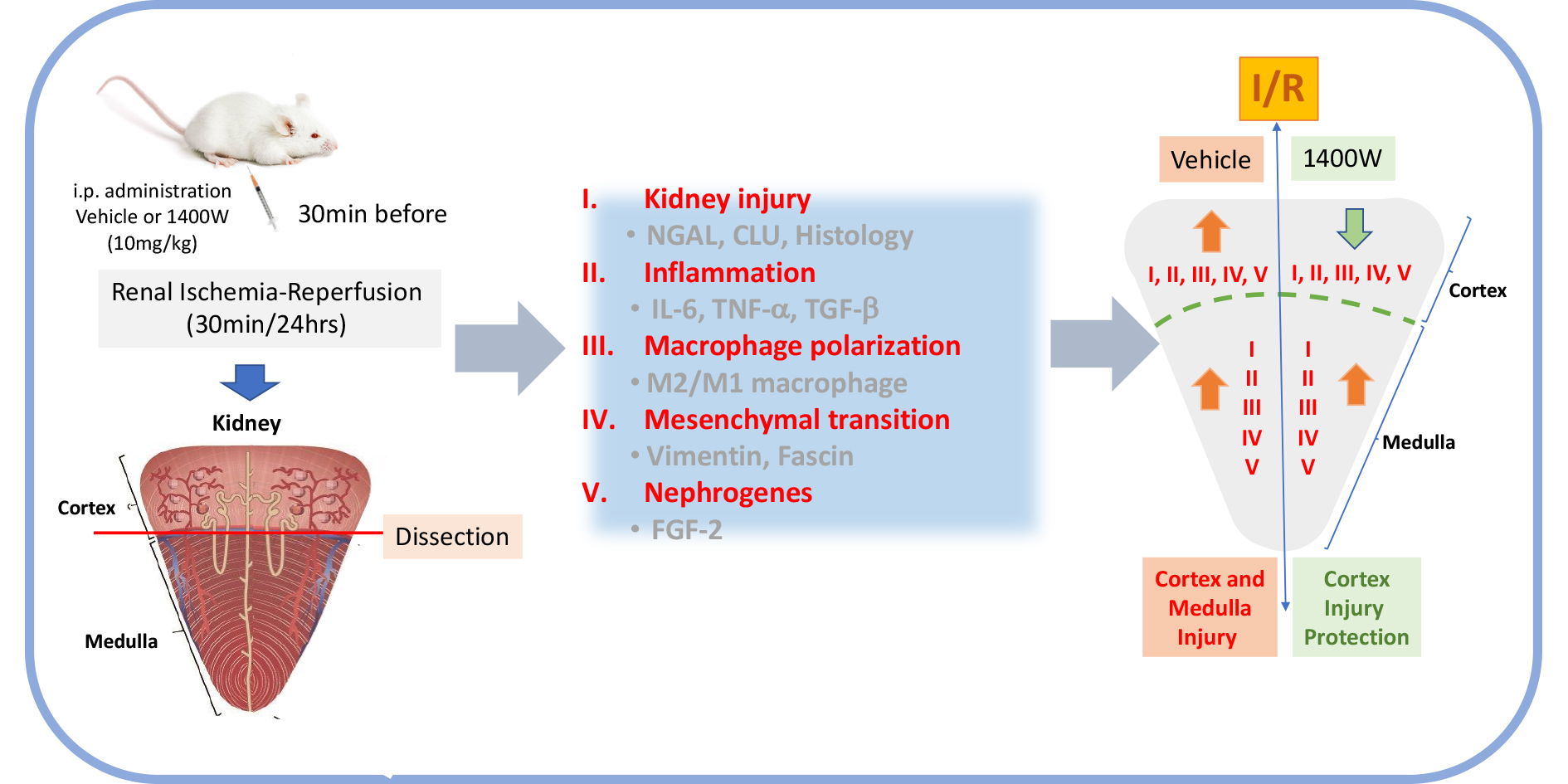
Acute kidney injury (AKI) is a group of syndromes defined by an abrupt decrease in glomerular filtration and is associated with considerable morbidity, mortality, and high costs [1]. Renal ischemia-reperfusion (I/R) injury is a significant cause of AKI, which is triggered by a transient reduction of blood flow followed by blood reperfusion. I/R injury can lead to acute cell death, tissue injury, and renal dysfunction [2, 3]. Renal I/R is involved in transitioning from AKI to chronic kidney disease (CKD) [4]. The clinical intervention addressed fluid management, but there are no specific therapies for each type of AKI [5]. Effective drugs to protect patients against the renal damage induced by ischemia and reperfusion are still lacking. Therefore, research is needed to find effective drugs to prevent and treat AKI.
The nitric oxide (NO) produced by iNOS provokes kidney tissue injury because NO combines with the superoxide radical and forms the cytotoxic metabolite, peroxynitrite, which causes cell membrane damage through protein nitration. Thus, the iNOS inhibition should ameliorate kidney damage [6-10]. Previously, we demonstrated that the pharmacological inhibition of iNOS with L-NIL (L-N(6)-(1-iminoethyl)lysine), a selective inhibitor of iNOS enzymatic activity, decreases the signs of renal damage induced by renal I/R in mice, reducing the oxidative stress and inflammatory pathway (TLR4 and IL-1β) [6]. Recently, we showed that aminoguanidine (AG), another iNOS inhibitor, protects the kidney injury induced by renal I/R in mice. AG recovered the GSH/GSSG ratio, the GST activity, and lipoperoxidation, preventing iNOS and Hsp27 upregulation. Moreover, AG inhibits the inflammation markers (IL-6, FOXP3, and IL-10 mRNA) upregulation [7]. In addition, other investigations have demonstrated that inhibiting the iNOS activity with specific iNOS inhibitors reduces oxidative stress, renal injury, and kidney dysfunction provoked by I/R or sepsis [8-10]. Moreover, iNOS knock-out animals are more resistant to kidney damage elicited by I/R than their wild-type counterparts [11]. Thus, the current information shows that iNOS inhibition prevents oxidative stress, inflammation, and kidney dysfunction observed by I/R. Therefore, it is necessary to increase the evidence to establish iNOS as a molecular target to prevent and treat AKI in humans. In this paper, we bet on the inhibitor 1400W, which has been reported to be low toxicity and is much more specific than other iNOS inhibitors [12-14]. Thus, in a model of cardiac I/R injury, 1400W (20 mg/kg) inhibited the NO production in mice treated with lipopolysaccharide (LPS) [15]. Besides, in a rat model of liver transplantation, the production of reactive nitrogen species (RNS), necrosis, and apoptosis was blocked by 1400W (5 µM) [16]. In addition, 1400W (20 mg/kg, rats) significantly reduced the volume of ischemic brain lesions, attenuated weight loss, and neurological dysfunction [17]. Besides,1400W (10 mg/kg) also reduced oxidative stress and the ischemia-reperfusion injury in an ex-vivo porcine donation model after the kidney donor’s circulatory death [18]. Moreover, a comparative study using melatonin (a powerful antioxidant, iNOS inhibitor, and a scavenger of peroxynitrite) and 1400W observed that both melatonin and 1400W (10 mg/kg) were efficient in ameliorating experimental I/R injury, including oxidative and nitrosative stress in kidneys. Moreover, melatonin was more effective than 1400W, possibly through scavenging free oxygen radicals and peroxynitrite [19]. In the present study, we explore the protective effect of 1400W against I/R separately in the renal cortex and medulla.
During the I/R injury, the tubular epithelial cells experience acute tubular necrosis (ATN) and activate the regeneration process against cell damage [20]. Epithelial cells of the proximal convoluted tubule undergo a sequence of events, including cell dedifferentiation and proliferation, followed by cell migration, redifferentiation, and repairing the damaged epithelium [21, 22]. Interestingly, during the repair process, re-expression of proteins that participate during kidney embryonic development (nephrogenes) has been observed [23-25]. These proteins include, among others, mesenchymal factors, such as vimentin [6, 7], fascin-1, and FGF-2 [25, 26]. Moreover, we previously found that AG treatment before ischemia/reperfusion significantly prevented the vimentin and fascin-1 upregulation induced by I/R [7].
On another side, I/R triggers inflammation and leukocyte infiltration inside the tubulointerstitial compartment [27]. The proinflammatory macrophages (M1 phenotype) increase after 1 hour of reperfusion, peaking at 24 hours and persisting for 7 days in mice kidneys. In contrast, M2 macrophages are present about 3-5 days after the initial injury, inducing cell proliferation and tissue repair in the kidney by secreting anti-inflammatory cytokines [28-30]. Interestingly, several immune cells express iNOS, among them macrophages, dendritic cells, and NK cells [31]. Although NO derived from iNOS is harmful in the kidney, an interesting work demonstrated that iNOS deficient mice exhibited enhanced M1 macrophage polarization without significant effects on M2 macrophages. Additionally, the iNOS inhibitor L-NIL significantly enhanced M1 macrophage polarization in vitro , suggesting that iNOS deficiency results in more severe inflammation [32]. Another study using a rat model of neuropathic pain showed that the treatment with 1400W (20 mg/kg) increased the plasma concentration of anti-inflammatory cytokines (IL-10) and pro-inflammatory cytokines (IL-1α, and IL-1β), suggesting that 1400W could alter the balance between pro- and anti-inflammatory cytokines [33]. Thus, the effect of 1400W in renal I/R is not clearly understood.
In this work, we investigated the role of 1400W in kidney injury, inflammation, macrophage polarization, mesenchymal transition, and nephrogenes separately in the renal cortex and medulla in a mice model of I/R. Remarkably, 1400W treatment reduce the I/R-activation signs of renal damage (tissue morphology, NGAL, and Clusterin expression), mesenchymal transition (vimentin and fascin-1), inflammation, macrophage polarization (M2/M1), and nephrogenes (FGF-2) in the renal cortex but not in the renal medulla. Therefore, the present investigation provides relevant information to propose to 1400W as an element for a therapeutic approach in AKI treatment.
Materials and Methods
Animals
Male Balb/c mice (20-25g and 2 months old) were housed in a 12 h light/dark cycle. Animals had food and water ad libitum and were maintained at the University de los Andes-Animal Care Facility [6, 7, 34]. All experimental procedures were in accordance with institutional and international standards for the humane care and use of laboratory animals (Animal Welfare Assurance Publication A5427-01, Office for Protection from Research Risks, Division of Animal Welfare. The National Institutes of Health). All procedures were approved by the Committee on the Ethics of Animal Experiments of the Universidad de los Andes, Chile.
Ischemia-reperfusion (I/R)
The animals were anesthetized with a volatile anesthetic (sevoflurane) and maintained on a 37°C blanket during the surgical procedure. A flank incision exposed both kidneys, and the renal pedicle was occluded for 30 minutes with a non-traumatic vascular clamp (cat N° 18055-02 Fine Science Tools). Renal blood flow was re-established (reperfusion phase) by clamp removal, and both incisions were sutured. Sham animals did not undergo renal pedicle occlusion [6, 7]. Mice were treated intraperitoneally (i.p.) with either vehicle (physiological saline) or 10mg/kg of 1400W [18, 19] (from MedChem Express, catalog HY-18731). Then, the animals were subjected to 24 hours of reperfusion.
Real-Time PCR
The experimental process was carried out according to how we describe before [35]. In brief, total RNA was isolated using a RNeasy Mini Kit (Cat Nº: 74104, Qiagen) according to the manufacturer’s directions. Extracted RNA was quantified at 260 nm in a Spectrophotometer (NanoDrop One, Thermo Scientific), and the RNA’s integrity (28S/18S ratio) was assessed by agarose gel electrophoresis. cDNA was prepared from total RNA (1,0 μg) using a Improm-IITM Reverse Transcription System (Cat Nº: A3800, Promega) and random hexamers primers. Then, PCR was performed duplicated for each experiment (Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (Cat Nº: 600882, Agilent). Amplicons were detected for Real-Time Fluorescence Detection (Rotor-Gene Q, Qiagen). The primers used are detailed in Table 1. Relative mRNA expression of the target genes was calculated using the −2∆∆ Ct method after normalization to the levels of 18S.
Morphological studies
Mice were anesthetized, as mentioned before, and kidneys were removed after tying the renal pedicle and then cut by a sagittal section in two halves, fixed in 10% formalin, included in paraffin, sectioned, dewaxed, rehydrated, and rinsed in water. After the pieces were dehydrated, they were embedded in paraffin, cut into 4-nm sections, mounted on glass slides, and stained with Hematoxylin and eosin (H/E) and Periodic Acid-Schiff (PAS) performed for light microscopy analysis. Morphological changes were analyzed blindly by a histologist (co-author).
Statistical analysis
Differences between groups were analyzed using the non-parametric Kruskal-Wallis ANOVA and posthoc Tukey test using GraphPad Prism Software. The level of significance was set at p < 0.05.
Results
Effect of 1400W on kidney injury during renal ischemia and reperfusion
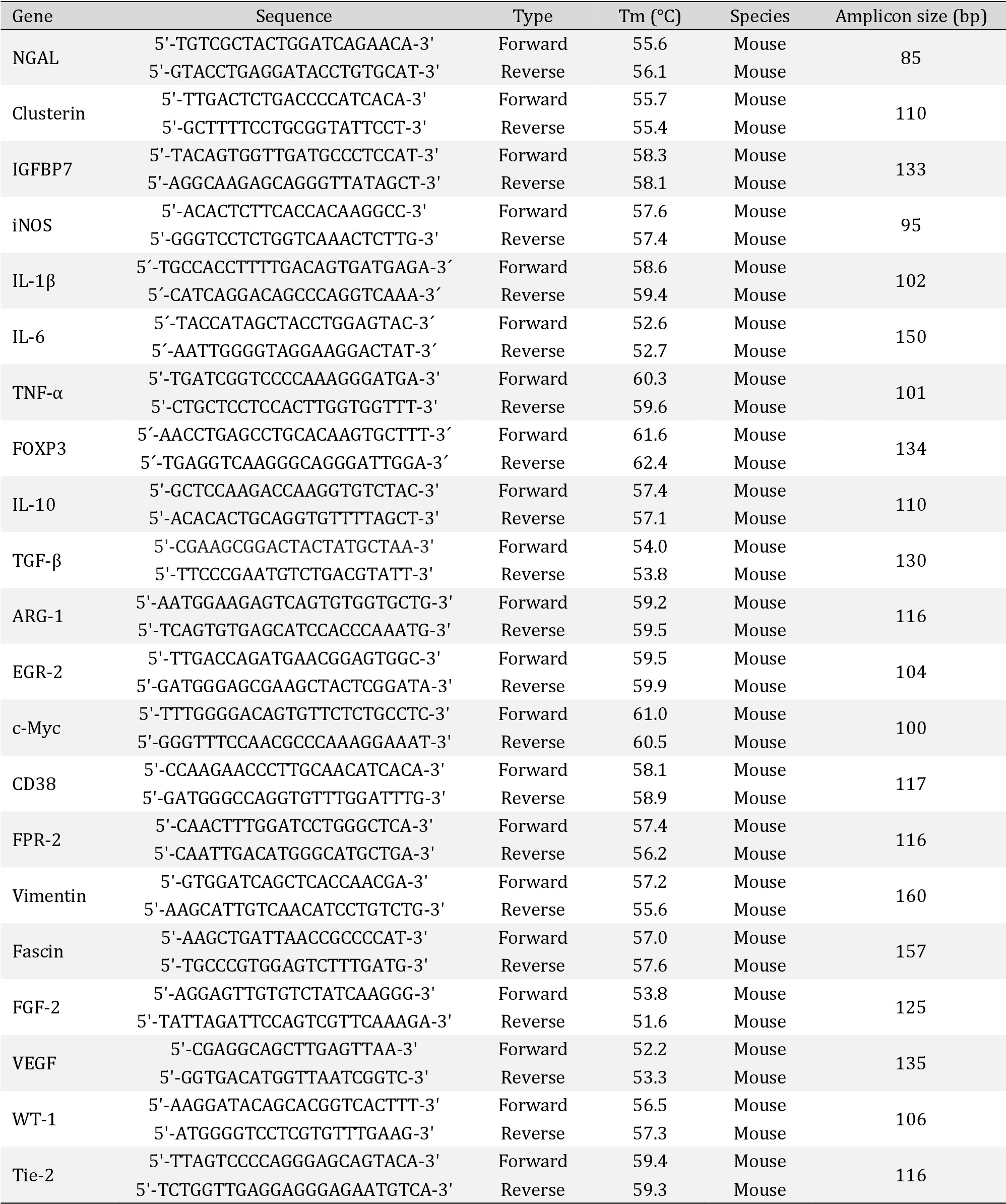
Kidneys from Balb/c adult mice were subjected to 30 minutes of ischemia and 24 hours of reperfusion. The kidney injury biomarker panel (NGAL, Clusterin, and IGFBP7) was assessed through mRNA expression in the kidney cortex and medulla separately. Compared with sham, I/R increased the NGAL and Clusterin mRNA expression in both kidney sections (cortex and medulla). Interestingly, 1400W decreased the NGAL and Clusterin I/R-upregulation only in the renal cortex but not in the renal medulla (Fig. 1A-B). The insulin-like growth factor-binding protein 7 (IGFBP7), a biomarker of risk of acute kidney injury [36] did not experiment change in the mRNA expression by I/R or 1400W (Fig. 1C). Additionally, we did not see changes in the iNOS mRNA expression by I/R or 1400W in the renal cortex and medulla (Fig. 1D).
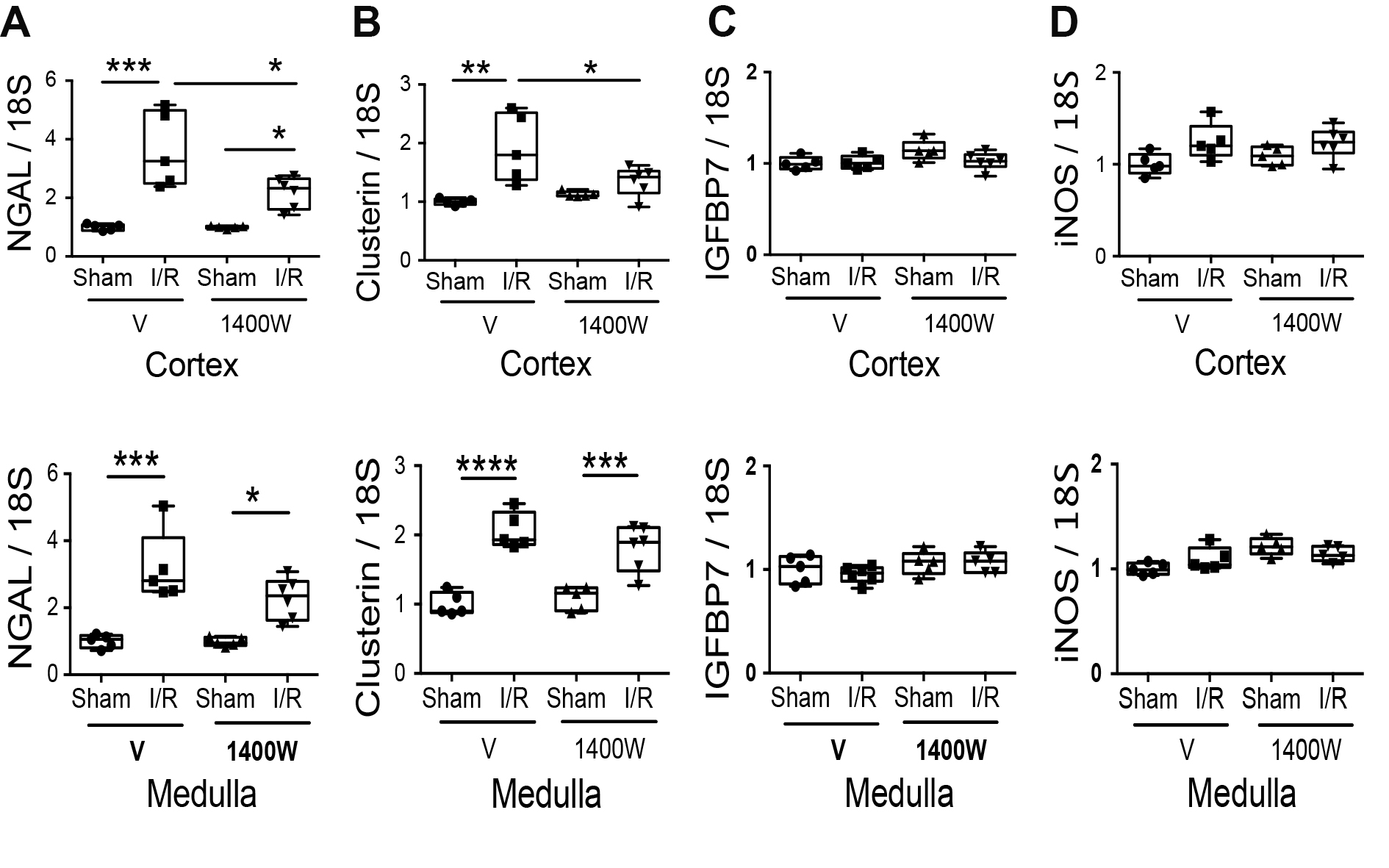
To improve the understanding of the effect of 1400W on kidney injury, we performed histology analysis. The I/R protocol did not produce morphological changes in the glomeruli (Fig. 2A). However, I/R provoked acute tubular injury in the renal cortex (Fig. 2B) and medulla (Fig. 2C). Compared with their respective sham group, the renal cortex in I/R mice showed acute tubular injury characterized by epithelial cell necrosis, flattening of the epithelium, and secondary distension leading to ectasia of tubular protein. In contrast, the pharmacological treatment with 1400W prevented these morphological alterations in the renal cortex. Only a thin area of subcapsular necrosis and cytoplasmic thick resorption droplets at proximal segments was observed (Fig. 2B). However, in the renal medulla, 1400W did not prevent extensive cytoplasmic flattening, tubular distension, sloughed epithelial cells with intratubular debris, and reabsorption of protein droplets at the straight proximal segments (Fig. 2C, arrows). The sham animals with saline or 1400W were normal (Fig. 2A, B, and C).
Effect of 1400W on inflammation during renal I/R
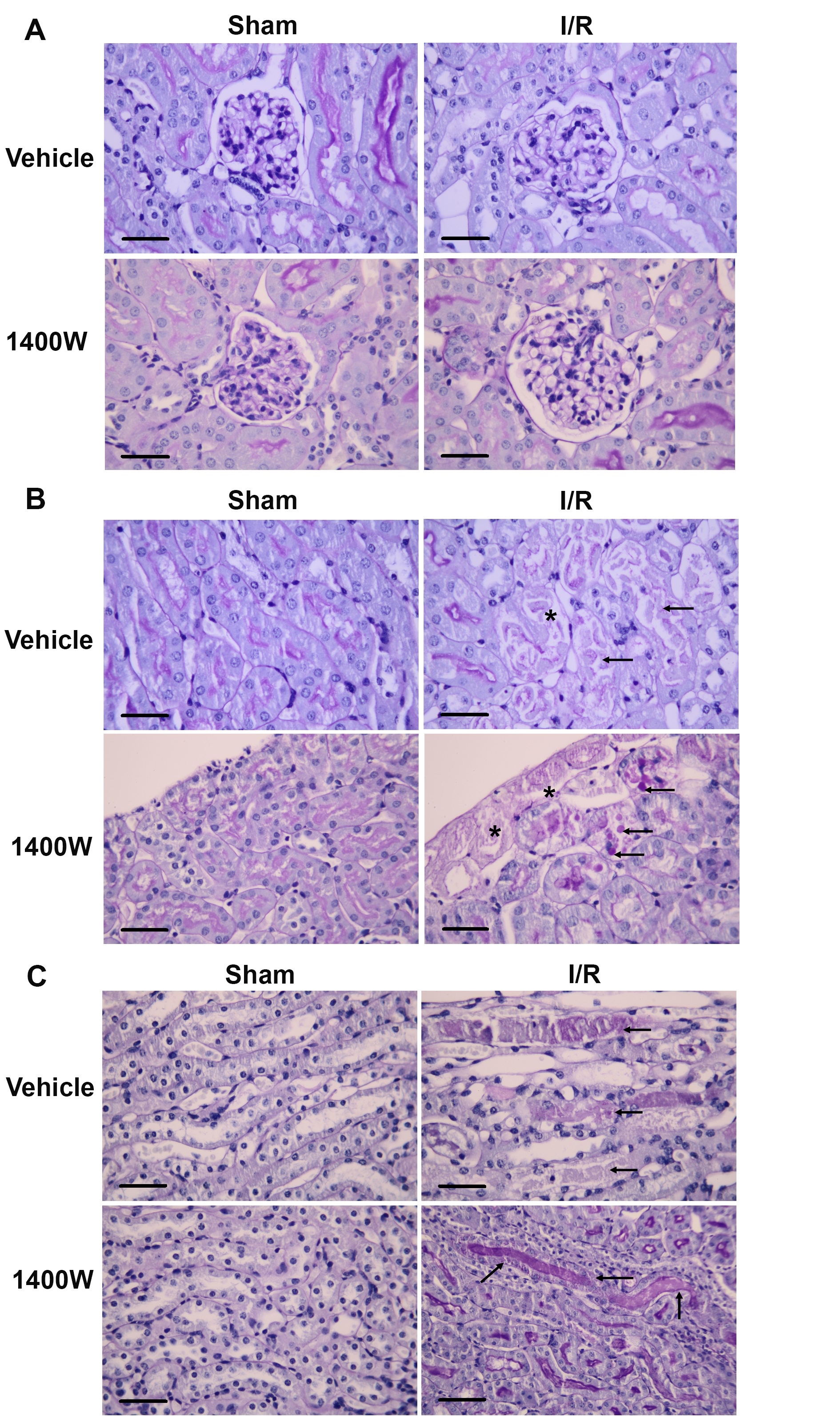
It has been widely demonstrated that I/R injury is associated with tubulointerstitial inflammation and exacerbates renal injury [21, 22]. Here, we studied the mRNA expression of a panel of inflammatory cytokines (IL-1β, IL-6, and TNF-α). We observed a significant upregulation of IL-6 and TNF-α mRNA in the renal cortex and medulla compared to the sham group (Fig. 3B-C). Interestingly, the I/R-upregulation of mRNA levels of IL-6 and TNF-α were prevented by 1400W pretreatment in the renal cortex but not in the renal medulla (Fig. 3B-C).
In contrast, the mRNA level of IL-1β was not modified at all (Fig. 3A). In addition, we studied the levels of anti-inflammatory markers (IL-10, Foxp3, and TGF-β). We did not find detectable levels of mRNA expression of IL-10 in the studied groups (data not shown). The Foxp3 mRNA levels were not modified by I/R or 1400W after 24 hours of reperfusion (Fig. 3D). However, the TGF-β mRNA level was upregulated in the renal cortex and medulla by I/R. Interestingly, the 1400W prevented the I/R-upregulation of mRNA levels of TGF-β in the renal cortex but not in the renal medulla (Fig. 3E). TGF-β signaling can induce M2 macrophage polarization in acute damage [37, 38]. Thus, we study the effect of 1400W in the M2 and M1 macrophage polarization in the experimental renal I/R model.
ABAB
Effect of 1400W on macrophage polarization during renal I/R
Macrophage polarization is a critical step in regulating inflammation during renal ischemia [29, 30]. We tested a panel of M2 (ArgI, Erg-2, and c-Myc) and M1 (Fpr-2 and CD38) macrophage markers to study the effect of 1400W in the I/R-induced macrophage polarization. As shown in Fig. 4A-C, the expression of M2 markers was significantly upregulated in the I/R group in both kidney regions (cortex and medulla) (except for ArgI in the renal medulla). The 1400W treatment prevented I/R-upregulation only in the renal cortex and not in the renal medulla. On another side, the M1 marker (Fpr-2, but not CD38) was significantly increased in the I/R mice in the renal cortex and medulla compared to the sham. The treatment with 1400W prevented the I/R-upregulation of Fpr-2 in the renal cortex and medulla. To better understand the effect of 1400W in the balance of M1 and M2 macrophage polarization, we analyzed the M2/M1 ([ArgI*Erg-2*c-Myc]/[Fpr-2*CD38]) ratio in each mouse. We observed that I/R significantly upregulated the M2/M1 ratio in the cortex and medulla. Consistent with the previous findings, the 1400W treatment avoided the I/R-upregulation of the M2/M1 ratio in the cortex but not in the medulla, suggesting that 1400W inhibited the inflammatory response in the renal cortex (Fig. 4F). Interestingly, in the renal medulla, 1400W prevented the M1 polarization without alterations in the M2 polarization of macrophages, suggesting that 1400W did not avoid the promotion of the kidney repair process in the medullary kidney under ischemia and reperfusion injury.
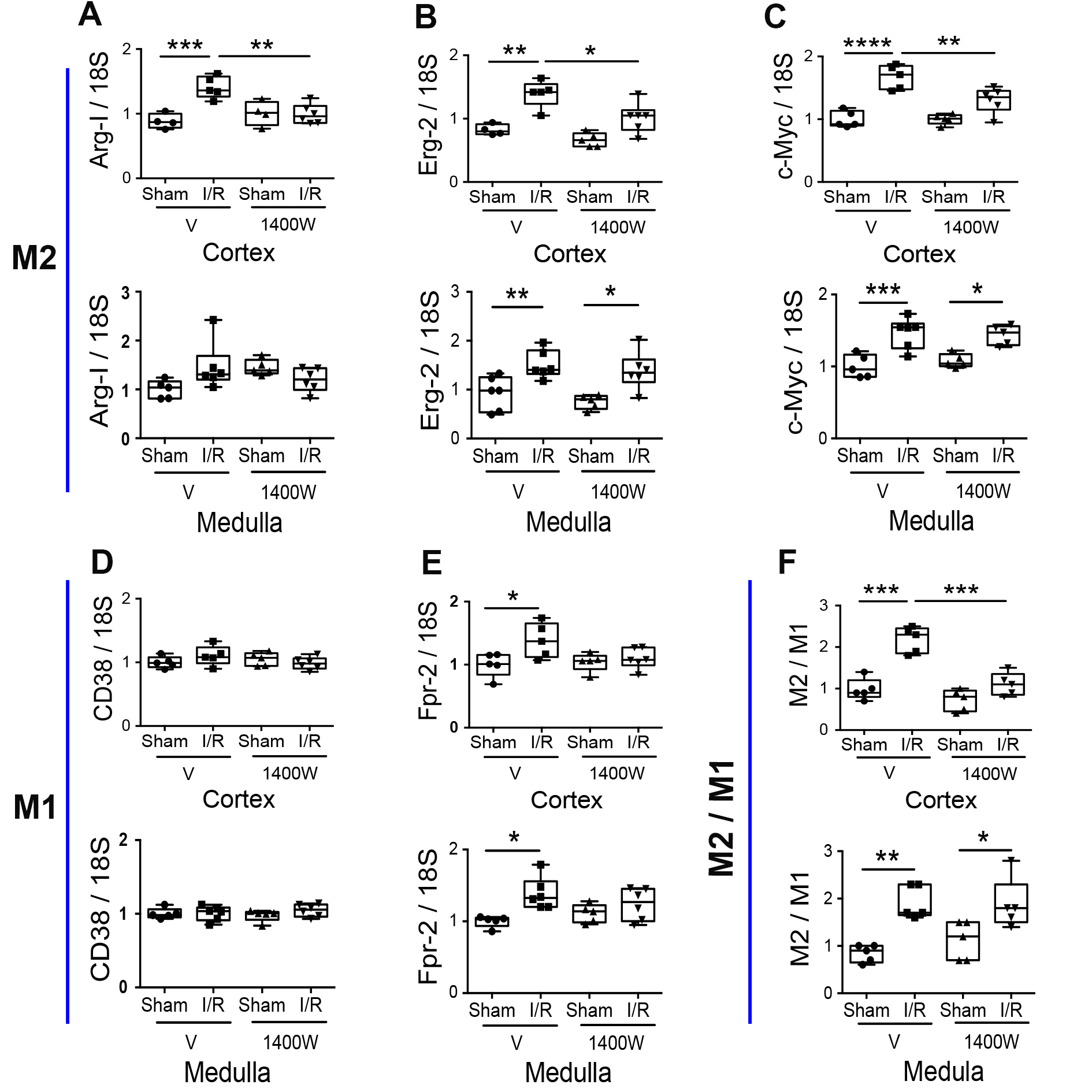
Effect of 1400W on mesenchymal markers expression during renal I/R
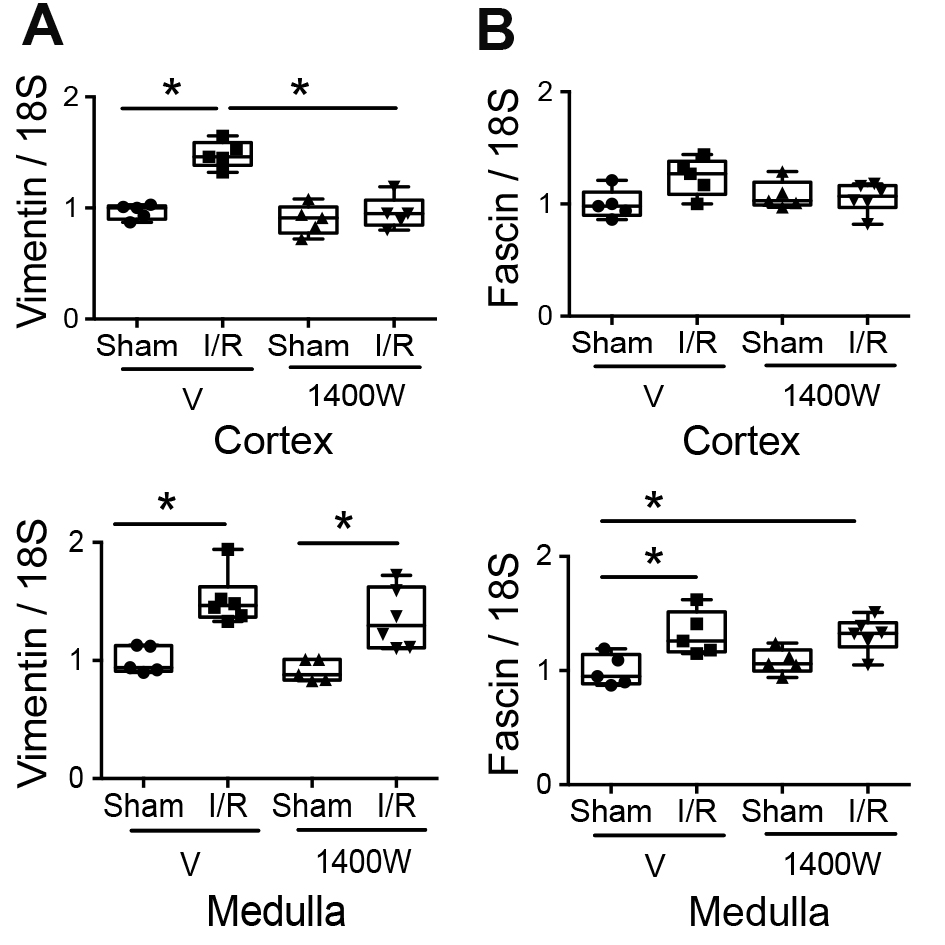
Vimentin and fascin-1 are expressed in the mesenchymal phenotype [6, 7]. Here, we found that the mRNA expression of Vimentin was upregulated in the cortex and medulla by I/R (Fig. 5A). Notably, 1400W treatment significantly prevented the renal I/R-induced upregulation of Vimentin only in the renal cortex. In addition, fascin-1 was upregulated only in the kidney medulla section of mice subjected to I/R stimulus, and it was not modified by 1400W treatment (Fig. 5B). These data suggested that 1400W prevented the I/R-upregulation of mesenchymal markers in the renal cortex but not in the renal medulla.
Effect of 1400W on nephrogenic gene expression during renal I/R
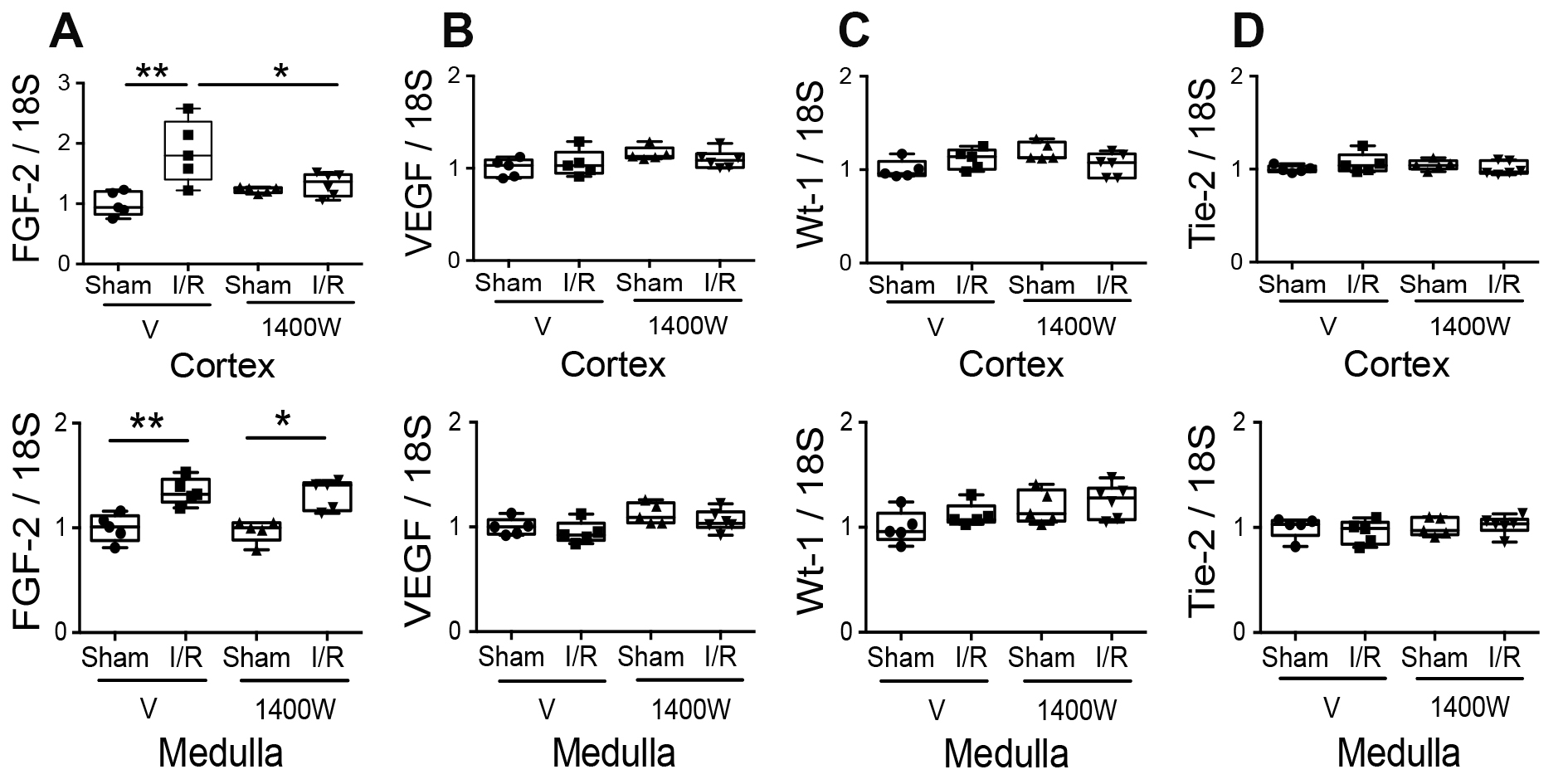
On another side, Fibroblast Growth Factor 2 (FGF-2) is a protein secreted during kidney development but is less expressed in adult kidney epithelial cells. However, it is reexpressed in response to damage induced by I/R [25, 26] and attenuates I/R Injury via inhibition of endoplasmic reticulum stress [39]. Our results showed that FGF-2 was upregulated in the renal cortex and medulla in animals exposed to I/R damage compared to the respective sham group. The 1400W treatment before I/R significantly decreased FGF-2 expression in the renal cortex but not in the renal medulla (Fig. 6A). Our group previously described the expression of other nephrogenic proteins in tubular cells after kidney damage induced by I/R in rats [25]. They are reexpressed after I/R, such as the vascular endothelial growth factor (VEGF), the angiopoietin receptor (Tie-2), and the Wilms’ tumor gene (WT-1), a transcription factor that induces the transformation of mesenchymal cells into metanephrogenic tissue during kidney embryology. In our experimental I/R model, we did not detect changes in the mRNA of these genes after I/R or 1400W treatment with 24 hours of reperfusion (Fig. 6B-D).
Altogether, these results show clear signs of damage, inflammation, macrophage polarization, mesenchymal transition, and nephrogenes reexpression in the renal cortex and medulla induced by I/R injury. Remarkable, the pharmacological inhibition of iNOS using 1400W prevented the I/R-induced kidney alterations in the renal cortex but not in the renal medulla.
The assistance of the Animal Care and Veterinary of Nataly Quezada from Services of the Universidad de los Andes is gratefully acknowledged. The technical support of the histology technician Miguel Vargas from Laboratorio Inmunocel, also is gratefully acknowledged.
Author Contributions
Conceived and designed experiments: CEI, CP. Performed the experiments: CP, ML, GM. Analyzed the data: CP, ML, GM, CEI. Wrote the paper CP, CEI. Contributed reagents/materials/analysis tools: CP, GM, CEI.
Funding Sources
This study was supported by a grant from: FAI-Puente (FONDECYT-Iniciación). Universidad de los Andes-Consuelo Pasten and FAI-Universidad de los Andes- Carlos E. Irarrázabal.
Statement of Ethics
All experimental procedures were under institutional and international standards for humane care and laboratory animal use (Animal Welfare Assurance Publication A5427-01 Office for Protection from Research Risks, Division of Animal Welfare. NIH, USA). All procedures were approved by the Committee on the Ethics of Animal Experiments of the University de los Andes, Chile.
The authors have no conflicts of interest to declare.
| 1 Bellomo R, Kellum JA, Ronco C: Acute kidney injury. Lancet 2012;380:756-766. https://doi.org/10.1016/S0140-6736(11)61454-2 |
||||
| 2 Dong Y, Zhang Q, Wen J, Chen T, He L, Wang Y, Yin J, Wu R, Xue R, Li S, Fan Y, Wang N: Ischemic Duration and Frequency Determines AKI-to-CKD Progression Monitored by Dynamic Changes of Tubular Biomarkers in IRI Mice. Front Physiol 2019;10:153. https://doi.org/10.3389/fphys.2019.00153 |
||||
| 3 Khadzhynov D, Schmidt D, Hardt J, Rauch G, Gocke P, Eckardt KU, Schmidt-Ott KM: The Incidence of Acute Kidney Injury and Associated Hospital Mortality. Dtsch Arztebl Int 2019;116:397-404. https://doi.org/10.3238/arztebl.2019.0397 |
||||
| 4 Liu J, Kumar S, Dolzhenko E, Alvarado GF, Guo J, Lu C, Chen Y, Li M, Dessing MC, Parvez RK, Cippà PE, Krautzberger AM, Saribekyan G, Smith AD, McMahon AP: Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017;2:e94716. https://doi.org/10.1172/jci.insight.94716 |
||||
| 5 Perner A, Prowle J, Joannidis M, Young P, Hjortrup PB, Pettilä V: Fluid management in acute kidney injury. Intensive Care Med 2017;43:807-815. https://doi.org/10.1007/s00134-017-4817-x |
||||
| 6 Pasten C, Alvarado C, Rocco J, Contreras L, Aracena P, Liberona J, Suazo C, Michea L, Irarrázabal CE: l-NIL prevents the ischemia and reperfusion injury involving TLR-4, GST, clusterin, and NFAT-5 in mice. Am J Physiol Renal Physiol 2019;316:F624-F634. https://doi.org/10.1152/ajprenal.00398.2018 |
||||
| 7 Pasten C, Lozano M, Rocco J, Carrión F, Alvarado C, Liberona J, Michea L, Irarrázabal CE: Aminoguanidine Prevents the Oxidative Stress, Inhibiting Elements of Inflammation, Endothelial Activation, Mesenchymal Markers, and Confers a Renoprotective Effect in Renal Ischemia and Reperfusion Injury. Antioxidants (Basel) 2021;10:1724. https://doi.org/10.3390/antiox10111724 |
||||
| 8 Noiri E, Peresleni T, Miller F, Goligorsky MS: In vivo targeting of inducible NO synthase with oligodeoxynucleotides protects rat kidney against ischemia. J Clin Invest 1996;97:2377-2383. https://doi.org/10.1172/JCI118681 |
||||
| 9 Chatterjee PK, Patel NS, Kvale EO, Cuzzocrea S, Brown PA, Stewart KN, Mota-Filipe H, Thiemermann C: Inhibition of inducible nitric oxide synthase reduces renal ischemia/reperfusion injury. Kidney Int 2002;61:862-871. https://doi.org/10.1046/j.1523-1755.2002.00234.x |
||||
| 10 Chatterjee PK, Patel NS, Sivarajah A, Kvale EO, Dugo L, Cuzzocrea S, Brown PAJ, Stewart KN, Mota-Filipe H, Britti D, Yaqoob MM, Thiemermann C: GW274150, a potent and highly selective inhibitor of iNOS, reduces experimental renal ischemia/reperfusion injury. Kidney Int 2003;63:853-865. https://doi.org/10.1046/j.1523-1755.2003.00802.x |
||||
| 11 Ling H, Edelstein C, Gengaro P, Meng X, Lucia S, Knotek M, Wangsiripaisan A, Shi Y, Schrier R: Attenuation of renal ischemia-reperfusion injury in inducible nitric oxide synthase knockout mice mice. Am J Physiol Ren Physiol 1999;277:F383-F390. https://doi.org/10.1152/ajprenal.1999.277.3.F383 |
||||
| 12 Cinelli MA, Do HT, Miley GP, Silverman RB: Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med Res Rev 2020;40:158-189. https://doi.org/10.1002/med.21599 |
||||
| 13 Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG: 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo . J Biol Chem 1997; 272:4959-4963. https://doi.org/10.1074/jbc.272.8.4959 |
||||
| 14 Boer R, Ulrich WR, Klein T, Mirau B, Haas S, Baur I: The Inhibitory Potency and Selectivity of Arginine Substrate Site Nitric-Oxide Synthase Inhibitors Is Solely Determined by Their Affinity toward the Different Isoenzymes. Molecular Pharmacology 2000;58:1026-1034. https://doi.org/10.1124/mol.58.5.1026 |
||||
| 15 Wang E, Feng Y, Zhang M, Zou L, Li Y, Buys ES, Huang P, Brouckaert P, Chao W: Toll-Like Receptor 4 Signaling Confers Cardiac Protection Against Ischemic Injury via Inducible Nitric Oxide Synthase- and Soluble Guanylate Cyclase-Dependent Mechanisms. Anesthesiology 2011;114:603-613. https://doi.org/10.1097/ALN.0b013e31820a4d5b |
||||
| 16 Shi Y, Rehman H, Wright GL, Zhong Z: Inhibition of inducible nitric oxide synthase prevents graft injury after transplantation of livers from rats after cardiac death. Liver Transpl 2010;16:1267-1277. https://doi.org/10.1002/lt.22148 |
||||
| 17 Parmentier S, Böhme GA, Lerouet D, Damour D, Stutzmann JM, Margaill I, Plotkine M: Selective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. Br J Pharmacol 1999;127:546-552 https://doi.org/10.1038/sj.bjp.0702549 |
||||
| 18 Hosgood SA, Yates PJ, Nicholson ML: 1400W reduces ischemia reperfusion injury in an ex-vivo porcine model of the donation after circulatory death kidney donor. World J Transplant 2014;4:299-305. https://doi.org/10.5500/wjt.v4.i4.299 |
||||
| 19 Ersoz N, Guven A, Cayci T, Uysal B, Turk E, Oztas E, Akgul EO, Korkmaz A, Cetiner S: Comparison of the efficacy of melatonin and 1400W on renal ischemia/reperfusion injury: a role for inhibiting iNOS. Ren Fail 2009;31:704-710. https://doi.org/10.3109/08860220903085989 |
||||
| 20 Legrand M, Mik EG, Johannes T, Payen D, Ince C: Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol Med 2008;14:502-516. https://doi.org/10.2119/2008-00006.Legrand |
||||
| 21 Kumar S: Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int 2018;93:27-40. https://doi.org/10.1016/j.kint.2017.07.030 |
||||
| 22 Rayego-Mateos S, Marquez-Expósito L, Rodrigues-Diez R, Sanz AB, Guiteras R, Doladé N, Rubio-Soto I, Manonelles A, Codina S, Ortiz A, Cruzado JM, Ruiz-Ortega M, Sola A: Molecular Mechanisms of Kidney Injury and Repair. Int J Mol Sci 2022; 23:1542. https://doi.org/10.3390/ijms23031542 |
||||
| 23 Devarajan P, Mishra J, Supavekin S, Patterson LT, Steven Potter S: Gene expression in early ischemic renal injury: clues towards pathogenesis, biomarker discovery, and novel therapeutics. Mol Genet Metab 2003;80:365-376. https://doi.org/10.1016/j.ymgme.2003.09.012 |
||||
| 24 Rudman-Melnick V, Adam M, Potter A, Chokshi SM, Ma Q, Drake KA, Schuh MP, Kofron JM, Devarajan P, Potter SS: Single-Cell Profiling of AKI in a Murine Model Reveals Novel Transcriptional Signatures, Profibrotic Phenotype, and Epithelial-to-Stromal Crosstalk. J Am Soc Nephrol 2020;31:2793-2814. https://doi.org/10.1681/ASN.2020010052 |
||||
| 25 Villanueva S, Céspedes C, Vio CP: Ischemic acute renal failure induces the expression of a wide range of nephrogenic proteins. Am J Physiol Regul Integr Comp Physiol 2006;290:R861-R870. https://doi.org/10.1152/ajpregu.00384.2005 |
||||
| 26 Little MH, Kairath P: Does Renal Repair Recapitulate Kidney Development? J Am Soc Nephrol 2017;28:34-46. https://doi.org/10.1681/ASN.2016070748 |
||||
| 27 Patel AA, Ginhoux F, Yona S: Monocytes, macrophages, dendritic cells, and neutrophils: an update on lifespan kinetics in health and disease. Immunology 2021;163:250-261. https://doi.org/10.1111/imm.13320 |
||||
| 28 Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG: Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 2011;22:317-326. https://doi.org/10.1681/ASN.2009060615 |
||||
| 29 Xie X, Yang X, Wu J, Ma J, Wei W, Fei X, Wang M: Trib1 Contributes to Recovery From Ischemia/Reperfusion-Induced Acute Kidney Injury by Regulating the Polarization of Renal Macrophages. Front Immunol 2020;11:473. https://doi.org/10.3389/fimmu.2020.00473 |
||||
| 30 Ma S, Wang DH: Knockout of Trpa1 Exacerbates Renal Ischemia-Reperfusion Injury With Classical Activation of Macrophages. Am J Hypertens 2021;34:110-116. https://doi.org/10.1093/ajh/hpaa162 |
||||
| 31 Xue Q, Yan Y, Zhang R, Xiong H: Regulation of iNOS on Immune Cells and Its Role in Diseases. Int J Mol Sci 2018;19:3805. https://doi.org/10.3390/ijms19123805 |
||||
| 32 Lu G, Zhang R, Geng S, Peng L, Jayaraman P, Chen C, Xu F, Yang J, Li Q, Zheng H, Shen K, Wang J, Liu X, Wang W, Zheng Z, Qi CF, Si C, He JC, Liu K, Lira SA, et al.: Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat Commun 2015;6:6676. https://doi.org/10.1038/ncomms7676 |
||||
| 33 Staunton CA, Barrett-Jolley R, Djouhri L, Thippeswamy T: Inducible nitric oxide synthase inhibition by 1400W limits pain hypersensitivity in a neuropathic pain rat model. Exp Physiol 2018;103:535-544. https://doi.org/10.1113/EP086764 |
||||
| 34 Pasten C, Herrera-Luna Y, Lozano M, Rocco J, Alvarado C, Liberona J, Michea L, Irarrázabal CE: Glutathione S-Transferase and Clusterin, New Players in the Ischemic Preconditioning Renal Protection in a Murine Model of Ischemia and Reperfusion. Cell Physiol Biochem 2021;55:635-650. https://doi.org/10.33594/000000442 |
||||
| 35 Serman Y, Fuentealba RA, Pasten C, Rocco J, Ko BCB, Carrión F, Irarrázabal CE: Emerging new role of NFAT5 in inducible nitric oxide synthase in response to hypoxia in mouse embryonic fibroblast cells. Am J Physiol Cell Physiol 2019;317:C31-C38. https://doi.org/10.1152/ajpcell.00054.2019 |
||||
| 36 Meersch M, Schmidt C, Van Aken H, Rossaint J, Görlich D, Stege D, Malec E, Januszewska K, Zarbock A: Validation of cell-cycle arrest biomarkers for acute kidney injury after pediatric cardiac surgery. PLoS One 2014;9:e110865. https://doi.org/10.1371/journal.pone.0110865 |
||||
| 37 Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, Wang H, Fang R, Bu X, Cai S, Du J: TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016;7:52294-52306. https://doi.org/10.18632/oncotarget.10561 |
||||
| 38 Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N: TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol 2012;13:31. https://doi.org/10.1186/1471-2172-13-31 |
||||
| 39 Tan X, Tao Q, Li G, Xiang L, Zheng X, Zhang T, Wu C, Li D: Fibroblast Growth Factor 2 Attenuates Renal Ischemia-Reperfusion Injury via Inhibition of Endoplasmic Reticulum Stress. Front Cell Dev Biol 2020;8:147. https://doi.org/10.3389/fcell.2020.00147 |
||||
| 40 Koike Y, Yozaki M, Utani A, Murota H: Fibroblast growth factor 2 accelerates the epithelial-mesenchymal transition in keratinocytes during wound healing process. Sci. Rep 2020;10:1-13. https://doi.org/10.1038/s41598-020-75584-7 |
||||
| 41 Villanueva S, Cespedes C, Gonzalez A, Vio CP: bFGF induces an earlier expression of nephrogenic proteins after ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol 2006;291:R1677-1687. https://doi.org/10.1152/ajpregu.00023.2006 |
||||
| 42 Villanueva S, Cespedes C, Gonzalez AA, Roessler E, Vio CP: Inhibition of bFGF-receptor type 2 increases kidney damage and suppresses nephrogenic protein expression after ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol 2008;294:R819-R828. https://doi.org/10.1152/ajpregu.00273.2007 |
||||
| 43 Deng LC, Alinejad T, Bellusci S, Zhang JS: Fibroblast Growth Factors in the Management of Acute Kidney Injury Following Ischemia-Reperfusion. Front Pharmacol 2020;11:426. https://doi.org/10.3389/fphar.2020.00426 |
||||
| 44 Pyrillou K, Burzynski LC, Clarke MCH: Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front Immunol 2020;11:613170. https://doi.org/10.3389/fimmu.2020.613170 |
||||
| 45 Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ: Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int 2009;75:511-517. https://doi.org/10.1038/ki.2008.600 |
||||
| 46 Ray SC, Mason J, O'Connor PM: Ischemic Renal Injury: Can Renal Anatomy and Associated Vascular Congestion Explain Why the Medulla and Not the Cortex Is Where the Trouble Starts? Semin Nephrol 2019;39:520-529. https://doi.org/10.1016/j.semnephrol.2019.10.002 |
||||
| 47 Crislip GR, O'Connor PM, Wei Q, Sullivan JC: Vasa recta pericyte density is negatively associated with vascular congestion in the renal medulla following ischemia reperfusion in rats. Am J Physiol Renal Physiol 2017;313:F1097-F1105. https://doi.org/10.1152/ajprenal.00261.2017 |
||||
| 48 Ray SC, Sun J, O'Connor P: Ischemia-induced vascular congestion of the ascending vasa recta precedes congestion of the peritubular capillaries in the renal medulla. The FASEB Journal 2020; DOI: . https://doi.org/10.1096/fasebj.2020.34.s1.04767 |
||||
| 49 Freitas F, Attwell D: Pericyte-mediated constriction of renal capillaries evokes no-reflow and kidney injury following ischaemia. Elife 2022;11:e74211. https://doi.org/10.7554/eLife.74211 |
||||
| 50 Shahid M, Francis J, Majid DS: Tumor necrosis factor-alpha induces renal vasoconstriction as well as natriuresis in mice. Am J Physiol Renal Physiol 2008;295:F1836-F1844. https://doi.org/10.1152/ajprenal.90297.2008 |
||||
| 51 Kim BS and Goligorsky MS: Role of VEGF in kidney development, microvascular maintenance, and pathophysiology of renal disease. Korean J Intern Med 2003;18:65-75. https://doi.org/10.3904/kjim.2003.18.2.65 |
||||
| 52 Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P: Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol 1998;141:1659-1673. https://doi.org/10.1083/jcb.141.7.1659 |
||||
| 53 Zhang F, Siow YL, O K: Hyperhomocysteinemia activates NF-kappaB and inducible nitric oxide synthase in the kidney. Kidney Int 2004;65:1327-1338. https://doi.org/10.1111/j.1523-1755.2004.00510.x |
||||
| 54 Radi R, Beckman JS, Bush KM, Freeman BA: Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 1991;288:481-487. https://doi.org/10.1016/0003-9861(91)90224-7 |
||||
| 55 Kocak-Toker N, Giris M, Tülübas F, Uysal M, Aykac-Toker G: Peroxynitrite induced decrease in Na+, K+-ATPase activity is restored by taurine. World J Gastroenterol 2005;11:3554-3557. https://doi.org/10.3748/wjg.v11.i23.3554 |
||||
| 56 Dasgupta S, Gomez JJ, Singh I, Khan M: S-Nitrosylation in Regulation of Inflammation and Cell Damage. Curr Drug Targets 2018;19:1831-1838. https://doi.org/10.2174/1389450119666180213094747 |
||||