×
![]()
Corresponding Author: Ben D. Perry
School of Science, Western Sydney University, Narellan Rd & Gilchrist Dr, Campbelltown NSW 2560 (Australia)
Tel. +61 2 4620 3276, E-Mail b.perry@westernsydney.edu.au
Skeletal Muscle and Kidney Crosstalk in Chronic Kidney Disease
Kayte A. Jenkin Ben D. Perry
School of Science, Western Sydney University, Sydney, Australia
Introduction
The functioning of complex organisms requires a constant and delicate balance of processes both between and within cells, tissues, and organ systems. Our understanding of how hormones and cytokines can affect function in different organ systems has been pivotal to elucidating the physiology and pathophysiology behind many processes [1, 2]. There is growing appreciation for the role of signalling crosstalk connecting different organ systems of the body, even from tissues traditionally classified as “inert” in terms of their capacity to produce chemical signals that can act on other organ systems [3-5]. These bioactive proteins are often classified and grouped based on the tissue producing them - adipokines produced by adipose tissue, hepatokines from liver, and myokines from muscle. Essentially, many organs have much more of a functional interaction with other systems than what we once thought was possible.
Many of these secreted molecules contribute to, or can exacerbate, a variety of functions and diseases in other organ systems, even if the two organs are not traditionally considered as having a linked or shared function [6]. For example, there is a strong association with skeletal muscle atrophy and muscle dysfunction in patients with chronic kidney disease (CKD) [7, 8]. Inversely, patients with CKD who are able to maintain muscle mass and continue habitual exercise and can reduce disease progression and prevent renal function decline [9, 10]. Considering the large volume of blood filtered by the kidneys, and the substantial metabolic role and relative mass of skeletal muscle, it is unsurprising that there is evidence of physiological crosstalk between skeletal muscle and kidney. To date, research has primarily examined the role of kidney dysfunction on muscle atrophy via indirect factors such as metabolic acidosis [11], chronic inflammation [12], and impaired insulin signalling [8]. Muscle atrophy caused by metabolic acidosis present in CKD is perhaps one of the most well-understood examples of kidney-muscle cross talk. Metabolic acidosis is a milieu commonly associated with CKD, as the kidneys are unable to maintain their capacity to secrete endogenous acid generated from metabolic processes, resulting in an overall increase in hydrogen ion concentration in the body. Progressive impairment of kidney function causes metabolic acidosis, which subsequently drives muscle atrophy primarily through upregulation of muscle protein degradation via the suppression of the IRS-PI3K-Akt insulin signalling pathway, which subsequently increases FoxO transcription
activity [8]. The IRS-PI3K-Akt pathway is not only responsible for insulin-mediated glucose uptake via GLUT4 (Glucose transporter type 4) in skeletal muscle, but also plays a pivotal role in protein degradation. The phosphorylation of Akt (Protein kinase B) causes subsequent phosphorylation of FoxO (Forkhead box class O), which excludes FoxO from the nucleus, decreasing its transcriptional activity of E3 ubiquitin ligases. If activation of the IRS-PI3K-Akt pathway is decreased, FoxO activity is subsequently increased, which stimulates transcription of E3 ubiquitin ligases including Atrogin-1 and MuRF-1 (Muscle Ring Factor-1) for targeting of proteins to be degraded by the ubiquitin proteasome system [7, 8, 13]. In CKD, acidosis contributes to downregulation of the IRS1-PI3K-Akt pathway and subsequently increases ubiquitin-proteasome mediated protein degradation. Glucocorticoids also induce atrophy via decreasing PI3K (Phosphoinositide 3-kinase) activity, affecting both protein synthesis and degradation in skeletal muscle [14].
Whilst the relationship between muscle wasting and CKD is complex and multifactorial in nature, it is only recently that specific signalling molecules that directly contribute to this kidney-muscle relationship have been identified. Identification of molecules that are produced and secreted by skeletal muscle have existed for some time, and Pedersen, et al. [15] coined the term “myokines” for these autocrine, paracrine and endocrine signalling factors originating from skeletal muscle, with IL-6 (Interleukin 6) being one of the first examples [4, 15]. A decade later, emerging evidence started to link the ability of myokines to directly affect kidney functioning in the context of chronic kidney disease [16-19]. More recently, in various models of nephropathy it has been demonstrated that the kidneys themselves can produce cachectic factors which directly mediate atrophy in skeletal muscle [5]. This review will focus on crosstalk in both directions between skeletal muscle and the kidney. The interaction of these organs in pathological conditions like Chronic Kidney Disease (CKD) and muscle atrophy, sarcopenia, and cachexia will be examined, as well inter-organ crosstalk in a non-disease state, where the beneficial effects of exercise will be examined in terms of kidney function. For the purposes of this review, the term “crosstalk” will refer to direct secreted humeral factors from kidney or skeletal muscle, which affects the other organ system. Our understanding of how the kidneys and skeletal muscle interact with each other is key to elucidating the pathophysiology processes that drive both health and disease.
The Impact of CKD on Skeletal Muscle Mass and Function
Chronic kidney disease (CKD) is a common condition, affecting 10-15% of the worldwide adult population [20]. CKD is disease defined as abnormalities in kidney structure (podocyte loss, tubular hypertrophy, fibrosis) or function (reduced glomerular filtration rate, elevated proteinuria, creatinine or BUN; Blood Urea Nitrogen) for three months or more [21]. CKD severity is clinically assessed in five stages, with Stage 1 being normal glomerular filtration rate, and minor structural damage or elevation of urine markers, to Stage 5, which is considered end stage kidney failure. CKD is a serious disease that once it progresses to Stage 5, can be fatal if not treated by continuing dialysis or kidney transplant [20]. CKD is often associated with other comorbidities, including diabetes, hypertension, cardiomyopathy, muscle atrophy, and muscle dysfunction [8, 10, 16, 21-23]. Muscle wasting conditions can encompass a spectrum of symptoms, from a loss of skeletal muscle mass, termed muscle atrophy, or progressive muscle weakness and loss with aging known as sarcopenia, to a complex metabolic syndrome known as cachexia, where patients exhibit involuntary and pathological weight loss of more than 5% of their body weight over 12 months [24].
Reduced muscle function and reductions in lean muscle mass are common outcomes for people living with CKD. Symptoms and treatment of CKD contributes to alterations in both the catabolic and anabolic processes which are required to maintain muscle mass. It is estimated that between 11-28% of patients with CKD have sarcopenia [25] - although the incidence may be as high as 54% depending on clinical definitions, cut off points, and diagnostic tools used to gauge incidence, as rates vary between experimental models and regions [26]. As the disease progresses, each stage of CKD increases the risk of sarcopenia by an additional 45% [27]. Protein degradation in skeletal muscle can be via the activation of the ubiquitin-proteasome (UPS), lysosome-autophagy, and calpain systems. In CKD patients, a loss of lean muscle mass and reduced muscle function is clinically important, as muscle atrophy and cachexia are both associated with a higher mortality rate [28]. Primary skeletal muscle cells obtained from CKD patients retain their cachexia phenotype in vitro [29], and dialysis has been shown to directly alter protein metabolism, stimulating protein breakdown in skeletal muscle during dialysis treatment, with proteolytic processes persisting for 2-hours post treatment [30]. Patients with more advanced CKD are often directed by medical professionals to lower their dietary intake of protein (or may do so spontaneously) in order to combat intraglomerular pressure and hyper-filtration, to help maintain and preserve renal function. However, dietary protein restriction may also contribute to the development of muscle atrophy and sarcopenia [16, 31, 32]. The causes of muscle atrophy and cachexia in CKD are multifaceted with several factors contributing to protein degradation, including malnutrition, inflammation and acidosis and are reviewed elsewhere [16, 33]. The following sections of this review will the factors activin A (released by the kidney), myostatin, microRNA’s, irisin and mitsugumin 53 (released by muscle) during disease which have been shown to have a direct effect on the other organ.
Kidney-Derived Factors which Affect Muscle Mass and Function
Activin A
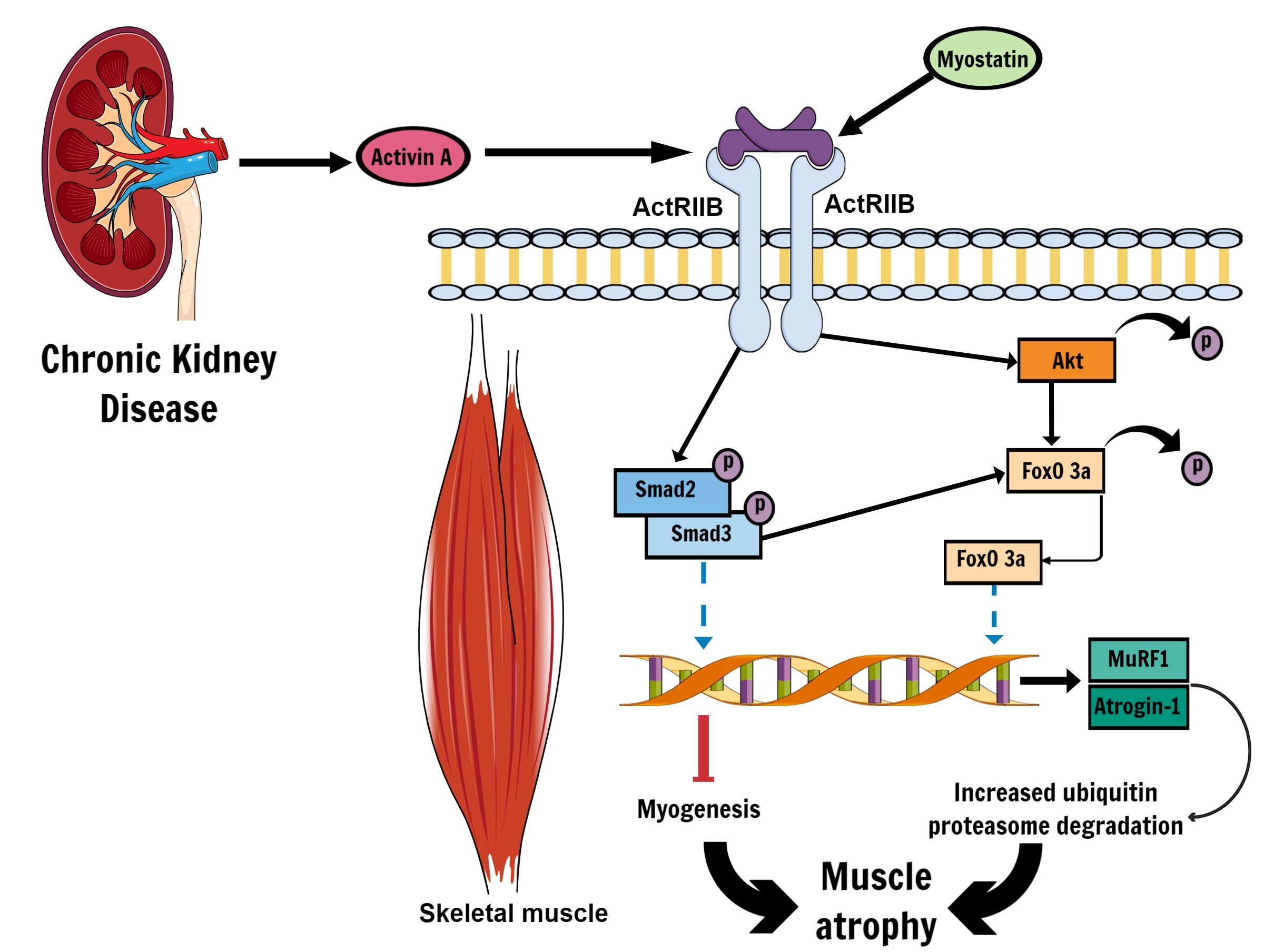
Activin A is a protein that was identified in the 1980s and characterised as an endogenous antagonist to the hormone inhibin [34]. Activin A belongs to the TGF-β (Transforming Growth Factor beta) superfamily of growth and differentiation factors, which elicits effects via two kinase receptors known as activin receptor II type A (ActRIIA) and B (ActRIIB) [34-37]. Overexpression of activin A can have detrimental effects on the normal structure and function of both the kidneys and skeletal muscle independently [35-41]. In the kidneys, overexpression of activin A during embryonic development can inhibit tubule and ureteric bud formation, and cause alterations in the proliferation and differentiation patterns of kidney cell populations via increased Pax-2 expression [40]. Indeed, inhibition of activin A via Follistatin reduced fibrosis caused by unilateral ureteral obstruction in rats [41]. In vitro and in vivo, healthy kidney expresses activin A at low levels, or is not detected at all [5, 37, 41]. In humans, circulating levels of activin A in blood serum is negatively correlated with estimated Glomerular Filtrations Rate (eGFR) and positively correlated with renal fibrosis [5]. In various models of nephropathy, activin A is an autocrine factor which causes the upregulation of fibrotic factors such as α-smooth muscle actin (α-SMA) and collagen types I and IV, and increases the proliferation and differentiation of renal fibroblast cells into myofibroblasts [5, 37].
In skeletal muscle, activation of ActRIIB by activin A (and other endogenous ligands such as myostatin, TGF-β, and BMP-11) will initiate phosphorylation of Smad2/Smad3 and pAKT-FoxO signalling cascade (Fig. 1) that leads to muscle wasting and cachexia via transcription of E3 ubiquitin ligases MuRF1 and Atrogin-1, which upregulates ubiquitin-proteasome mediated protein degradation [36, 38, 42]. Through both Smad2/Smad3 and pAKT-FoxO signalling activin A expression also mediates reduced skeletal muscle growth and myogenesis [38, 43], and inhibition of activin A has been shown to mitigate weight loss (total body weight and lean body weight), improve skeletal muscle mass, and help retain muscle strength in cancer cachexia [35, 36]. Promisingly, the activin A inhibitor Follistatin instigates muscle hypertrophy and may have utility in combating atrophy, especially if effects can be localised to skeletal muscle [44, 45]. However, as Follistatin can inhibit both myostatin and activin A [45], it is unclear to what extent the potential anti-atrophic effects of Follistatin are due to activin A inhibition alone. To the best of the authors’ knowledge, the effects of exogenous Follistatin has not been directly investigated in CKD-induced muscle atrophy and presents an intriguing future area of research.
There is emerging evidence of direct crosstalk between the kidney and skeletal muscle as a mechanism that promotes muscle atrophy in CKD. In comprehensive work recently published by Solagna, et al. [5], signalling cross-talk between skeletal muscle and kidneys was demonstrated using a number of different mouse models of nephropathy, and by examining cachexic factors of patients with CKD. In this study, activin A was shown to be near-exclusively produced by juxtaglomerular tubular cells and fibroblasts of the kidney in both human and rodent models of nephropathy, and higher levels of circulating activin A was positively correlated with worse outcomes for both kidney and skeletal muscle structure and function [5]. This work identified the role of kidney-derived activin A directly mediating muscle atrophy in a genetic mouse model (using the kidney-specific knockout of the kinase kif3a gene) and with a separate model of CKD, and demonstrated that pharmacologically blocking activin A in skeletal muscle also led to reduced renal fibrosis and improved renal function [5]. Pharmacological inhibition of activin A or blockade of activin A in skeletal muscle using an adenovirus vector in mice attenuated muscle mass loss, increased the cross-sectional area of muscle fibres, improved tetanic specific force and mitochondrial density in muscle fibres. The attenuation of muscle atrophy was shown to be mediated via a combination of increased protein expression via the mTOR pathway and the downregulation of FoXO-dependent gene transcription, which is involved in protein degradation. Simultaneously, inhibition of activin A in skeletal muscle alone improved kidney function and structure, highlighting that activin A crosstalk between kidneys and muscle is not unidirectional. Accumulation of activin A was also inversely associated with estimated GFR in CKD patients, suggesting that declining kidney function not only produced activin A, but also dampened the clearance of activin A. The emergence of this direct crosstalk mechanism between the kidneys and muscle is an exciting development and may be indicative that other kidney-derived factors, or “renalkines”, could have some direct role in muscle physiology in CKD and renal failure; although mechanistic studies in humans are required.
Author Contributions
BP and KJ wrote, proofread, prepared figures, and finalized the manuscript.
Funding Sources
The article processing costs for this review was funded by Western Sydney University.
Statement of Ethics
The authors have no ethical conflicts to disclose.
The authors have no conflicts of interest to declare.
| 1 Oppenheim JJ: Cytokines: past, present, and future. Int J Hematol 2001;74:3-8. https://doi.org/10.1007/BF02982543 |
||||
| 2 Tata JR: One hundred years of hormones. EMBO Rep 2005;6:490-496. https://doi.org/10.1038/sj.embor.7400444 |
||||
| 3 Romero A, Eckel J: Organ Crosstalk and the Modulation of Insulin Signaling. Cells 2021;10:2082. https://doi.org/10.3390/cells10082082 |
||||
| 4 Severinsen MCK, Pedersen BK: Muscle-organ crosstalk: the emerging roles of myokines. Endocr Rev 2020;41:594-609. https://doi.org/10.1210/endrev/bnaa016 |
||||
| 5 Solagna F, Tezze C, Lindenmeyer MT, Lu S, Wu G, Liu S, Zhao Y, Mitchell R, Meyer C, Omairi S: Pro-cachectic factors link experimental and human chronic kidney disease to skeletal muscle wasting programs. J Clin Invest 2021;131:e135821. https://doi.org/10.1172/JCI135821 |
||||
| 6 Senesi P, Luzi L, Terruzzi I: Adipokines, Myokines, and Cardiokines: The Role of Nutritional Interventions. Int J Mol Sci 2020;21:8372. https://doi.org/10.3390/ijms21218372 |
||||
| 7 Bailey JL, Zheng B, Hu Z, Price SR, Mitch WE: Chronic kidney disease causes defects in signaling through the insulin receptor substrate/phosphatidylinositol 3-kinase/Akt pathway: implications for muscle atrophy. J Am Soc Nephrol 2006;17:1388-1394. https://doi.org/10.1681/ASN.2004100842 |
||||
| 8 Price SR, Gooch JL, Donaldson SK, Roberts-Wilson TK: Muscle atrophy in chronic kidney disease results from abnormalities in insulin signaling. J Ren Nutr 2010;20:S24-S28. https://doi.org/10.1053/j.jrn.2010.05.007 |
||||
| 9 Roshanravan B, Gamboa J, Wilund K: Exercise and CKD: Skeletal Muscle Dysfunction and Practical Application of Exercise to Prevent and Treat Physical Impairments in CKD. Am J Kidney Dis 2017;69:837-852. https://doi.org/10.1053/j.ajkd.2017.01.051 |
||||
| 10 Wang B, Zhang C, Zhang A, Cai H, Price SR, Wang XH: MicroRNA-23a and microRNA-27a mimic exercise by ameliorating CKD-induced muscle atrophy. J Am Soc Nephrol 2017;28:2631-2640. https://doi.org/10.1681/ASN.2016111213 |
||||
| 11 Bailey JL, Wang X, England BK, Price SR, Ding X, Mitch WE: The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest 1996;97:1447-1453. https://doi.org/10.1172/JCI118566 |
||||
| 12 Cheung WW, Paik KH, Mak RH: Inflammation and cachexia in chronic kidney disease. Pediatr Nephrol 2010;25:711-724. https://doi.org/10.1007/s00467-009-1427-z |
||||
| 13 Franch HA, Raissi S, Wang X, Zheng B, Bailey JL, Price SR: Acidosis impairs insulin receptor substrate-1-associated phosphoinositide 3-kinase signaling in muscle cells: consequences on proteolysis. Am J Physiol Renal Physiol 2004;287:F700-F706. https://doi.org/10.1152/ajprenal.00440.2003 |
||||
| 14 Braun TP, Marks DL: The regulation of muscle mass by endogenous glucocorticoids. Front Physiol 2015;6:12. https://doi.org/10.3389/fphys.2015.00012 |
||||
| 15 Pedersen BK, Steensberg A, Fischer C, Keller C, Keller P, Plomgaard P, Febbraio M, Saltin B: Searching for the exercise factor: is IL-6 a candidate? J Muscle Res Cell Motil 2003;24:113-119. https://doi.org/10.1023/A:1026070911202 |
||||
| 16 Wang XH, Mitch WE: Mechanisms of muscle wasting in chronic kidney disease. Nat Rev Nephrol 2014;10:504-516. https://doi.org/10.1038/nrneph.2014.112 |
||||
| 17 Robinson KA, Baker LA, Graham-Brown MP, Watson EL: Skeletal muscle wasting in chronic kidney disease: the emerging role of microRNAs. Nephrol Dial Transplant 2020;35:1469-1478. https://doi.org/10.1093/ndt/gfz193 |
||||
| 18 Zhang A, Li M, Wang B, Klein JD, Price SR, Wang XH: miRNA‐23a/27a attenuates muscle atrophy and renal fibrosis through muscle‐kidney crosstalk. J Cachexia Sarcopenia Muscle 2018;9:755-770. https://doi.org/10.1002/jcsm.12296 |
||||
| 19 Zhang A, Wang H, Wang B, Yuan Y, Klein JD, Wang XH: Exogenous miR‐26a suppresses muscle wasting and renal fibrosis in obstructive kidney disease. FASEB J 2019;33:13590-13601. https://doi.org/10.1096/fj.201900884R |
||||
| 20 Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, Fox CS, Gansevoort RT, Heerspink HJL, Jardine M, Kasiske B, Kottgen A, Kretzler M, Levey AS, Luyckx VA, Mehta R, Moe O, Obrador G, Pannu N, Parikh CR, et al.: Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 2017;390:1888-1917. https://doi.org/10.1016/S0140-6736(17)30788-2 |
||||
| 21 Levey AS, Eckardt KU, Dorman NM, Christiansen SL, Hoorn EJ, Ingelfinger JR, Inker LA, Levin A, Mehrotra R, Palevsky PM, Perazella MA, Tong A, Allison SJ, Bockenhauer D, Briggs JP, Bromberg JS, Davenport A, Feldman HI, Fouque D, Gansevoort RT, et al.: Nomenclature for kidney function and disease: report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int 2020;97:1117-1129. https://doi.org/10.1016/j.kint.2020.02.010 |
||||
| 22 Peng H, Wang Q, Lou T, Qin J, Jung S, Shetty V, Li F, Wang Y, Feng XH, Mitch WE, Graham BH, Hu Z: Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat Commun 2017;8:1493. https://doi.org/10.1038/s41467-017-01646-6 |
||||
| 23 Wang B, Wang J, He W, Zhao Y, Zhang A, Liu Y, Hassounah F, Ma F, Klein JD, Wang XH: Exogenous miR-29a attenuates muscle atrophy and kidney fibrosis in unilateral ureteral obstruction mice. Hum Gene Ther 2020;31:367-375. https://doi.org/10.1089/hum.2019.287 |
||||
| 24 Koppe L, Fouque D, Kalantar-Zadeh K: Kidney cachexia or protein-energy wasting in chronic kidney disease: facts and numbers. J Cachexia Sarcopenia Muscle 2019;10:479-484. https://doi.org/10.1002/jcsm.12421 |
||||
| 25 Souza VA, Oliveira D, Barbosa SR, Correa J, Colugnati FAB, Mansur HN, Fernandes N, Bastos MG: Sarcopenia in patients with chronic kidney disease not yet on dialysis: Analysis of the prevalence and associated factors. PLoS One 2017;12:e0176230. https://doi.org/10.1371/journal.pone.0176230 |
||||
| 26 Chatzipetrou V, Begin MJ, Hars M, Trombetti A: Sarcopenia in Chronic Kidney Disease: A Scoping Review of Prevalence, Risk Factors, Association with Outcomes, and Treatment. Calcif Tissue Int 2022;110:1-31. https://doi.org/10.1007/s00223-021-00898-1 |
||||
| 27 Yu MD, Zhang HZ, Zhang Y, Yang SP, Lin M, Zhang YM, Wu JB, Hong FY, Chen WX: Relationship between chronic kidney disease and sarcopenia. Sci Rep 2021;11:20523. https://doi.org/10.1038/s41598-021-99592-3 |
||||
| 28 Noori N, Kopple JD, Kovesdy CP, Feroze U, Sim JJ, Murali SB, Luna A, Gomez M, Luna C, Bross R: Mid-arm muscle circumference and quality of life and survival in maintenance hemodialysis patients. Clin J Am Soc Nephrol 2010;5:2258-2268. https://doi.org/10.2215/CJN.02080310 |
||||
| 29 Baker LA, O'Sullivan TF, Robinson KA, Graham-Brown MPM, Major RW, Ashford RU, Smith AC, Philp A, Watson EL: Primary skeletal muscle cells from chronic kidney disease patients retain hallmarks of cachexia in vitro . J Cachexia Sarcopenia Muscle 2022;13:1238-1249. https://doi.org/10.1002/jcsm.12802 |
||||
| 30 Ikizler TA, Pupim LB, Brouillette JR, Levenhagen DK, Farmer K, Hakim RM, Flakoll PJ: Hemodialysis stimulates muscle and whole body protein loss and alters substrate oxidation. Am J Physiol Endocrinol Metab 2002;282:E107-116. https://doi.org/10.1152/ajpendo.2002.282.1.E107 |
||||
| 31 Herzog CA, Strief JW, Collins AJ, Gilbertson DT: Cause-specific mortality of dialysis patients after coronary revascularization: why don't dialysis patients have better survival after coronary intervention? Nephrol Dial Transplant 2008;23:2629-2633. https://doi.org/10.1093/ndt/gfn038 |
||||
| 32 Oosterwijk MM, Navis G, Bakker SJL, Laverman GD: Personalized Nutrition in Patients with Type 2 Diabetes and Chronic Kidney Disease: The Two-Edged Sword of Dietary Protein Intake. J Pers Med 2022;12:300. https://doi.org/10.3390/jpm12020300 |
||||
| 33 Wang XH, Mitch WE, Price SR: Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat Rev Nephrol 2022;18:138-152. https://doi.org/10.1038/s41581-021-00498-0 |
||||
| 34 Bloise E, Ciarmela P, Dela Cruz C, Luisi S, Petraglia F, Reis FM: Activin A in Mammalian Physiology. Physiol Rev 2019;99:739-780. https://doi.org/10.1152/physrev.00002.2018 |
||||
| 35 Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, Qian H, Lynch GS, Harrison CA, Gregorevic P: Elevated expression of activins promotes muscle wasting and cachexia. FASEB J 2014;28:1711-1723. https://doi.org/10.1096/fj.13-245894 |
||||
| 36 Walton KL, Chen JL, Arnold Q, Kelly E, La M, Lu L, Lovrecz G, Hagg A, Colgan TD, Qian H, Gregorevic P, Harrison CA: Activin A-Induced Cachectic Wasting Is Attenuated by Systemic Delivery of Its Cognate Propeptide in Male Mice. Endocrinology 2019;160:2417-2426. https://doi.org/10.1210/en.2019-00257 |
||||
| 37 Yamashita S, Maeshima A, Kojima I, Nojima Y: Activin A is a potent activator of renal interstitial fibroblasts. J Am Soc Nephrol 2004;15:91-101. https://doi.org/10.1097/01.ASN.0000103225.68136.E6 |
||||
| 38 Han HQ, Zhou X, Mitch WE, Goldberg AL: Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 2013;45:2333-2347. https://doi.org/10.1016/j.biocel.2013.05.019 |
||||
| 39 Lee SJ, Lehar A, Liu Y, Ly CH, Pham QM, Michaud M, Rydzik R, Youngstrom DW, Shen MM, Kaartinen V, Germain-Lee EL, Rando TA: Functional redundancy of type I and type II receptors in the regulation of skeletal muscle growth by myostatin and activin A. Proc Natl Acad Sci U S A 2020;117:30907-30917. https://doi.org/10.1073/pnas.2019263117 |
||||
| 40 Maeshima A, Maeshima K, Nojima Y, Kojima I: Involvement of Pax-2 in the action of activin A on tubular cell regeneration. J Am Soc Nephrol 2002;13:2850-2859. https://doi.org/10.1097/01.ASN.0000035086.93977.E9 |
||||
| 41 Maeshima A, Mishima K, Yamashita S, Nakasatomi M, Miya M, Sakurai N, Sakairi T, Ikeuchi H, Hiromura K, Hasegawa Y, Kojima I, Nojima Y: Follistatin, an activin antagonist, ameliorates renal interstitial fibrosis in a rat model of unilateral ureteral obstruction. Biomed Res Int 2014;2014:376191. https://doi.org/10.1155/2014/376191 |
||||
| 42 Bollinger LM, Witczak CA, Houmard JA, Brault JJ: SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner. Am J Physiol Cell Physiol 2014;307:C278-C287. https://doi.org/10.1152/ajpcell.00391.2013 |
||||
| 43 Loumaye A, Lause P, Zhong X, Zimmers TA, Bindels LB, Thissen J-P: Activin A Causes Muscle Atrophy through MEF2C-Dependent Impaired Myogenesis. Cells 2022;11:1119. https://doi.org/10.3390/cells11071119 |
||||
| 44 Castonguay R, Lachey J, Wallner S, Strand J, Liharska K, Watanabe AE, Cannell M, Davies MV, Sako D, Troy ME, Krishnan L, Mulivor AW, Li H, Keates S, Alexander MJ, Pearsall RS, Kumar R: Follistatin-288-Fc Fusion Protein Promotes Localized Growth of Skeletal Muscle. J Pharmacol Exp Ther 2019;368:435-445. https://doi.org/10.1124/jpet.118.252304 |
||||
| 45 Han X, Møller LLV, De Groote E, Bojsen-Møller KN, Davey J, Henríquez-Olguin C, Li Z, Knudsen JR, Jensen TE, Madsbad S, Gregorevic P, Richter EA, Sylow L: Mechanisms involved in follistatin-induced hypertrophy and increased insulin action in skeletal muscle. J Cachexia Sarcopenia Muscle 2019;10:1241-1257. https://doi.org/10.1002/jcsm.12474 |
||||
| 46 Verzola D, Barisione C, Picciotto D, Garibotto G, Koppe L: Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int 2019;95:506-517. https://doi.org/10.1016/j.kint.2018.10.010 |
||||
| 47 McPherron AC, Lawler AM, Lee SJ: Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997;387:83-90. https://doi.org/10.1038/387083a0 |
||||
| 48 Wang DT, Yang YJ, Huang RH, Zhang ZH, Lin X: Myostatin Activates the Ubiquitin-Proteasome and Autophagy-Lysosome Systems Contributing to Muscle Wasting in Chronic Kidney Disease. Oxid Med Cell Longev 2015;2015:684965. https://doi.org/10.1155/2015/684965 |
||||
| 49 Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M: Smad2 and 3 transcription factors control muscle mass in adulthood. American Journal of Physiology-Cell Physiology 2009;296:C1248-C1257. https://doi.org/10.1152/ajpcell.00104.2009 |
||||
| 50 Tando T, Hirayama A, Furukawa M, Sato Y, Kobayashi T, Funayama A, Kanaji A, Hao W, Watanabe R, Morita M, Oike T, Miyamoto K, Soga T, Nomura M, Yoshimura A, Tomita M, Matsumoto M, Nakamura M, Toyama Y, Miyamoto T: Smad2/3 Proteins Are Required for Immobilization-induced Skeletal Muscle Atrophy. J Biol Chem 2016;291:12184-12194. https://doi.org/10.1074/jbc.M115.680579 |
||||
| 51 Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, Song Y, Min H, Wang X, Du J, Mitch WE: Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 2011;25:1653-1663. https://doi.org/10.1096/fj.10-176917 |
||||
| 52 Verzola D, Procopio V, Sofia A, Villaggio B, Tarroni A, Bonanni A, Mannucci I, De Cian F, Gianetta E, Saffioti S, Garibotto G: Apoptosis and myostatin mRNA are upregulated in the skeletal muscle of patients with chronic kidney disease. Kidney Int 2011;79:773-782. https://doi.org/10.1038/ki.2010.494 |
||||
| 53 Yasar E, Tek NA, Tekbudak MY, Yurtdaş G, Gülbahar Ö, Uyar G, Ural Z, Çelik Ö M, Erten Y: The Relationship Between Myostatin, Inflammatory Markers, and Sarcopenia in Patients With Chronic Kidney Disease. J Ren Nutr 2022; DOI: 10.1053/j.jrn.2022.01.011. https://doi.org/10.1053/j.jrn.2022.01.011 |
||||
| 54 Rybalka E, Timpani CA, Debruin DA, Bagaric RM, Campelj DG, Hayes A: The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020;9:2657. https://doi.org/10.3390/cells9122657 |
||||
| 55 Yano S, Nagai A, Isomura M, Yamasaki M, Kijima T, Takeda M, Hamano T, Nabika T: Relationship between Blood Myostatin Levels and Kidney Function: Shimane CoHRE Study. PLoS One 2015;10:e0141035. https://doi.org/10.1371/journal.pone.0141035 |
||||
| 56 Delanaye P, Bataille S, Quinonez K, Buckinx F, Warling X, Krzesinski JM, Pottel H, Burtey S, Bruyere O, Cavalier E: Myostatin and Insulin-Like Growth Factor 1 Are Biomarkers of Muscle Strength, Muscle Mass, and Mortality in Patients on Hemodialysis. J Ren Nutr 2019;29:511-520. https://doi.org/10.1053/j.jrn.2018.11.010 |
||||
| 57 Zhou Y, Hellberg M, Hellmark T, Hoglund P, Clyne N: Muscle mass and plasma myostatin after exercise training: a substudy of Renal Exercise (RENEXC)-a randomized controlled trial. Nephrol Dial Transplant 2021;36:95-103. https://doi.org/10.1093/ndt/gfz210 |
||||
| 58 Palsgaard J, Brons C, Friedrichsen M, Dominguez H, Jensen M, Storgaard H, Spohr C, Torp-Pedersen C, Borup R, De Meyts P, Vaag A: Gene expression in skeletal muscle biopsies from people with type 2 diabetes and relatives: differential regulation of insulin signaling pathways. PLoS One 2009;4:e6575. https://doi.org/10.1371/journal.pone.0006575 |
||||
| 59 Verzola D, Milanesi S, Viazzi F, Ansaldo F, Saio M, Garibaldi S, Carta A, Costigliolo F, Salvidio G, Barisione C, Esposito P, Garibotto G, Picciotto D: Enhanced myostatin expression and signalling promote tubulointerstitial inflammation in diabetic nephropathy. Sci Rep 2020;10:6343. https://doi.org/10.1038/s41598-020-62875-2 |
||||
| 60 Wang H, Wang B, Zhang A, Hassounah F, Seow Y, Wood M, Ma F, Klein JD, Price SR, Wang XH: Exosome-mediated miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol Ther 2019;27:571-583. https://doi.org/10.1016/j.ymthe.2019.01.008 |
||||
| 61 Heiwe S, Jacobson SH: Exercise training in adults with CKD: a systematic review and meta-analysis. Am J Kidney Dis 2014;64:383-393. https://doi.org/10.1053/j.ajkd.2014.03.020 |
||||
| 62 Baker LA, March DS, Wilkinson TJ, Billany RE, Bishop NC, Castle EM, Chilcot J, Davies MD, Graham-Brown MPM, Greenwood SA, Junglee NA, Kanavaki AM, Lightfoot CJ, Macdonald JH, Rossetti GMK, Smith AC, Burton JO: Clinical practice guideline exercise and lifestyle in chronic kidney disease. BMC Nephrol 2022;23:75. https://doi.org/10.1186/s12882-021-02618-1 |
||||
| 63 Massart J, Sjogren RJO, Egan B, Garde C, Lindgren M, Gu W, Ferreira DMS, Katayama M, Ruas JL, Barres R, O'Gorman DJ, Zierath JR, Krook A: Endurance exercise training-responsive miR-19b-3p improves skeletal muscle glucose metabolism. Nat Commun 2021;12:5948. https://doi.org/10.1038/s41467-021-26095-0 |
||||
| 64 Gao HE, Li FH, Xie T, Ma S, Qiao YB, Wu DS, Sun L: Lifelong Exercise in Age Rats Improves Skeletal Muscle Function and MicroRNA Profile. Med Sci Sports Exerc 2021;53:1873. https://doi.org/10.1249/MSS.0000000000002661 |
||||
| 65 Mooren FC, Viereck J, Kruger K, Thum T: Circulating micrornas as potential biomarkers of aerobic exercise capacity. Am J Physiol-Heart C 2014;306:H557-H563. https://doi.org/10.1152/ajpheart.00711.2013 |
||||
| 66 Massart J, Sjogren RJO, Lundell LS, Mudry JM, Franck N, O'Gorman DJ, Egan B, Zierath JR, Krook A: Altered miR-29 Expression in Type 2 Diabetes Influences Glucose and Lipid Metabolism in Skeletal Muscle. Diabetes 2017;66:1807-1818. https://doi.org/10.2337/db17-0141 |
||||
| 67 Rodriguez-Carmona A, Perez Fontan M, Sangiao Alvarellos S, Garcia Falcon T, Pena Bello ML, Lopez Muniz A, Cordido F: Serum levels of the adipomyokine irisin in patients with chronic kidney disease. Nefrologia 2016;36:496-502. https://doi.org/10.1016/j.nefroe.2016.11.011 |
||||
| 68 Sadeghi Shad J, Akbari R, Qujeq D, Hajian-Tilaki K: Measurement of serum irisin in the different stages of chronic kidney disease. Caspian J Intern Med 2019;10:314-319. | ||||
| 69 Wen MS, Wang CY, Lin SL, Hung KC: Decrease in irisin in patients with chronic kidney disease. PLoS One 2013;8:e64025. https://doi.org/10.1371/journal.pone.0064025 |
||||
| 70 Flori L, Testai L, Calderone V: The "irisin system": From biological roles to pharmacological and nutraceutical perspectives. Life Sci 2021;267:118954. https://doi.org/10.1016/j.lfs.2020.118954 |
||||
| 71 Albrecht E, Norheim F, Thiede B, Holen T, Ohashi T, Schering L, Lee S, Brenmoehl J, Thomas S, Drevon CA, Erickson HP, Maak S: Irisin - a myth rather than an exercise-inducible myokine. Sci Rep 2015;5:8889. https://doi.org/10.1038/srep08889 |
||||
| 72 Cooke AB, Gomez YH, Daskalopoulou SS: 5 years later: irisin detection still an issue. Eur J Endocrinol 2017;177:C1-C4. https://doi.org/10.1530/EJE-17-0572 |
||||
| 73 Maak S, Norheim F, Drevon CA, Erickson HP: Progress and Challenges in the Biology of FNDC5 and Irisin. Endocr Rev 2021;42:436-456. https://doi.org/10.1210/endrev/bnab003 |
||||
| 74 Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, Nair KS, Gygi SP, Spiegelman BM: Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab 2015;22:734-740. https://doi.org/10.1016/j.cmet.2015.08.001 |
||||
| 75 Ma J, Chen K: The role of Irisin in multiorgan protection. Mol Biol Rep 2021;48:763-772. https://doi.org/10.1007/s11033-020-06067-1 |
||||
| 76 Timmons JA, Baar K, Davidsen PK, Atherton PJ: Is irisin a human exercise gene? Nature 2012;488:E9-10; discussion E10-11. https://doi.org/10.1038/nature11364 |
||||
| 77 Montes-Nieto R, Martinez-Garcia MA, Luque-Ramirez M, Escobar-Morreale HF: Differences in analytical and biological results between older and newer lots of a widely used irisin immunoassay question the validity of previous studies. Clin Chem Lab Med 2016;54:e199-201. https://doi.org/10.1515/cclm-2015-1071 |
||||
| 78 Whitson BA, Tan T, Gong N, Zhu H, Ma J: Muscle multiorgan crosstalk with MG53 as a myokine for tissue repair and regeneration. Curr Opin Pharmacol 2021;59:26-32. https://doi.org/10.1016/j.coph.2021.04.005 |
||||
| 79 Zhang Y, Wu HK, Lv F, Xiao RP: MG53: Biological Function and Potential as a Therapeutic Target. Mol Pharmacol 2017;92:211-218. https://doi.org/10.1124/mol.117.108241 |
||||
| 80 Duann P, Li H, Lin P, Tan T, Wang Z, Chen K, Zhou X, Gumpper K, Zhu H, Ludwig T, Mohler PJ, Rovin B, Abraham WT, Zeng C, Ma J: MG53-mediated cell membrane repair protects against acute kidney injury. Sci Transl Med 2015;7:279ra236. https://doi.org/10.1126/scitranslmed.3010755 |
||||
| 81 Li H, Duann P, Li Z, Zhou X, Ma J, Rovin BH, Lin PH: The cell membrane repair protein MG53 modulates transcription factor NF-kappaB signaling to control kidney fibrosis. Kidney Int 2022;101:119-130. https://doi.org/10.1016/j.kint.2021.09.027 |
||||
| 82 Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, Ko JK, Lin P, Thornton A, Zhao X, Pan Z, Komazaki S, Brotto M, Takeshima H, Ma J: MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol 2009;11:56-64. https://doi.org/10.1038/ncb1812 |
||||
| 83 Sermersheim M, Kenney AD, Lin PH, McMichael TM, Cai C, Gumpper K, Adesanya TMA, Li H, Zhou X, Park KH, Yount JS, Ma J: MG53 suppresses interferon-beta and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat Commun 2020;11:3624. https://doi.org/10.1038/s41467-020-17177-6 |
||||
| 84 Weisleder N, Takizawa N, Lin P, Wang X, Cao C, Zhang Y, Tan T, Ferrante C, Zhu H, Chen PJ, Yan R, Sterling M, Zhao X, Hwang M, Takeshima M, Cai C, Cheng H, Takeshima H, Xiao RP, Ma J: Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med 2012;4:139ra185. https://doi.org/10.1126/scitranslmed.3003921 |
||||
| 85 Tan T, Ko YG, Ma J: Dual function of MG53 in membrane repair and insulin signaling. BMB Rep 2016;49:414-423. https://doi.org/10.5483/BMBRep.2016.49.8.079 |
||||
| 86 Zhong W, Benissan-Messan DZ, Ma J, Cai C, Lee PHU: Cardiac effects and clinical applications of MG53. Cell Biosci 2021;11:115. https://doi.org/10.1186/s13578-021-00629-x |
||||
| 87 Bian Z, Wang Q, Zhou X, Tan T, Park KH, Kramer HF, McDougal A, Laping NJ, Kumar S, Adesanya TMA, Sermersheim M, Yi F, Wang X, Wu J, Gumpper K, Jiang Q, He D, Lin PH, Li H, Guan F, et al.: Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat Commun 2019;10:4659. https://doi.org/10.1038/s41467-019-12483-0 |
||||
| 88 Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, Kim BW, Lee H, Lee CS, Jeong BC, Song HK, Cho H, Kim YK, Lee JS, Park KS, Shin H, Choi I, Lee SH, Park WJ, Park SY, et al.: MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun 2013;4:2354. https://doi.org/10.1038/ncomms3354 |
||||
| 89 Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, Cao Y, Pi Y, Zhang X, Jin L, Zhang M, Jiang P, Liu F, Meng S, Zhang X, Jiang P, Cao CM, Xiao RP: Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 2013;494:375-379. https://doi.org/10.1038/nature11834 |
||||
| 90 Wu HK, Zhang Y, Cao CM, Hu X, Fang M, Yao Y, Jin L, Chen G, Jiang P, Zhang S, Song R, Peng W, Liu F, Guo J, Tang L, He Y, Shan D, Huang J, Zhou Z, Wang DW, et al.: Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019;139:901-914. https://doi.org/10.1161/CIRCULATIONAHA.118.037216 |
||||
| 91 Bianchi C, Raggi F, Rossi C, Frontoni S, Bonadonna RC, Del Prato S, Solini A: MG53 marks poor beta cell performance and predicts onset of type 2 diabetes in subjects with different degrees of glucose tolerance. Diabetes Metab 2021;48:101292. https://doi.org/10.1016/j.diabet.2021.101292 |
||||
| 92 Wang Q, Bian Z, Jiang Q, Wang X, Zhou X, Park KH, Hsueh W, Whitson BA, Haggard E, Li H, Chen K, Cai C, Tan T, Zhu H, Ma J: MG53 Does Not Manifest the Development of Diabetes in db/db Mice. Diabetes 2020;69:1052-1064. https://doi.org/10.2337/db19-0807 |
||||
| 93 Ma H, Liu J, Bian Z, Cui Y, Zhou X, Zhou X, Zhang B, Adesanya TM, Yi F, Park KH, Tan T, Chen Z, Zhu H: Effect of metabolic syndrome on mitsugumin 53 expression and function. PLoS One 2015;10:e0124128. https://doi.org/10.1371/journal.pone.0124128 |
||||