×
![]()
Corresponding Author: Carlos E. Irarrázabal
Programa de Fisiología, Laboratorio de Fisiología Integrativa y Molecular, Universidad de los Andes, S. Carlos Apoquindo 2200-Las Condes, Santiago (Chile)
Tel. +56-2-4129607, Fax +56-2-2141756 , E-Mail cirarrazabal@uandes.cl
The Ischemia and Reperfusion Injury Involves the Toll-Like Receptor-4 Participation Mainly in the Kidney Cortex
Yeimi Herrera-Lunaa Mauricio Lozanoa Consuelo Pastena,b Gabriele Multhoffc Carlos E. Irarrázabala,b
aLaboratorio de Fisiología Integrativa y Molecular, Programa de Fisiología, Centro de Investigación e Innovación Biomédica, Universidad de los Andes, Santiago, Chile, bEscuela de Medicina, Facultad de Medicina, Universidad de los Andes, Santiago, Chile, cDepartment of Radiation Oncology, Klinikum rechts der Isar, TU München (TUM), München, Germany
Introduction
The renal inflammatory response after ischemia and reperfusion injury (IRI) is associated with innate immunity activation, specifically with Toll-like receptors (TLRs), a family of proteins that function as critical mediators of innate immunity [1, 2]. As a result of injury, damaged, dead, or dying cells release damage-associated molecular patterns (DAMPs) activating TLR4 receptors. TLR4 signaling pathway operates via two distinct signaling pathways, namely MyD88 dependent and the MyD88 independent mechanisms. Both processes induce the transcription of proinflammatory cytokines by facilitating the nuclear translocation of NF-κB [2]. In addition, TLR4 also leads to upregulated macrophage infiltration and recruitment in kidneys during IRI [3]. TLR4 mediates the inflammatory activity and cytokine production associated with IRI in myocardial [4], cerebral [5], lung [6], and kidney [7].
The TLR4 is expressed basally on many kidney cells and is responsible to upregulates and promoting an influx of immune cells, such as dendritic, macrophages, and leukocyte cells into the damaged interstitium [7–10]. The role of TLR4 during IRI was studied previously using TLR4 knockout (TLR4-KO) animals. The TLR4-KO mice exposed to ischemia (45min) and reperfusion (1-10 days) had a partial reduction in chemokines levels, infiltrating granulocytes, renal damage, and improved renal function compared with WT mice [8]. In addition, mice deficient in TLR4 and MyD88 showed a partial protective effect against kidney dysfunction, tubular injury, neutrophil, macrophage accumulation, and expression of proinflammatory cytokines and chemokines [7, 10]. Another study showed that TLR4 deficient mice subjected to IRI (45min-24hrs reperfusion) had decreased levels of TNF-α, IL-1β, IL-6, IFN-γ, cell infiltration, apoptosis, and improvement of renal function compared with wild type mice [11]). Therefore, the literature indicated that TLR4 coordinates the innate immune response of the kidney against renal ischemia/reperfusion injury. Thus, targeting TLR4 could serve as a candidate for a therapeutic target to limit renal inflammation [2]. Conversely, TLR4 blockade during the healing phase suppressed IL-22 production and impaired kidney regeneration [12]. Therefore, it is necessary to improve the information to clarify the effect of TLR4 on the balance of injury and repair during renal IRI.
We studied the mesenchymal markers (vimentin and fascin), nitric oxidase synthase (iNOS), clusterin, heat shock proteins (Hsp27 and Hsp70), macrophage polarization (M1 and M2), and inflammatory factors (TNF-α, IL-1β, IL-6, IFN-γ, Foxp3, and IL-10) to better understand the role of TLR4 during IRI. Recently, was described that the mesenchymal markers fascin (an actin-bundling protein involved in cell motility) and vimentin (an intermediate filament, a complete mesenchymal cell phenotype marker) were upregulated in early posttransplant biopsies from allograft kidney with acute tubular necrosis and they play a detrimental role in long-term graft function [13]. Besides, the heat shock proteins (Hsp) have been considered important to protect against kidney injury. Thus, Hsp27 is a stress protein that shows an early and transient increase after acute ischemia, inhibiting apoptosis by decreasing intracellular reactive oxygen species and the mitochondrial caspase-dependent apoptotic pathway [14]. Hsp27 has a renoprotective function [15] and is a disease biomarker and therapeutic target [16]. In addition, the inducible Hsp70 family members are the most well-known proteins upregulated under stress. Their principal mode of action is by attenuation of cell death from apoptosis or necrosis. Previously was described that the renoprotective effect of Hsp70 is partially due to its direct immunomodulatory function on regulatory T cells (Tregs) [17]. The nuclear transcription factor Foxp3 (forkhead box P3) is a specific marker for CD4+CD25+ Tregs and regulates their development and function [18]. Furthermore, clusterin is a chaperone-like glycoprotein and promotes pro-survival autophagy [19], and its expression is upregulated after IRI, expressed primarily in the S3 segment and the distal tubule with distinct staining patterns in each segment [20]. Interestingly, clusterin deficiency worsens renal inflammation and tissue fibrosis after IRI in the kidney [21] and is required for renal tissue regeneration in the kidney repair phase after IRI, which is associated with the promotion of tubular cell proliferation [22]. The iNOS increases by IRI and has detrimental effects on the kidney by IRI [23, 24]. We previously published that iNOS and clusterin increase in the murine model of IRI [23, 25] and the pharmacological inhibition of iNOS (L-NIL), before IRI, improves renal function and prevents clusterin upregulation after 48hrs of reperfusion [23].
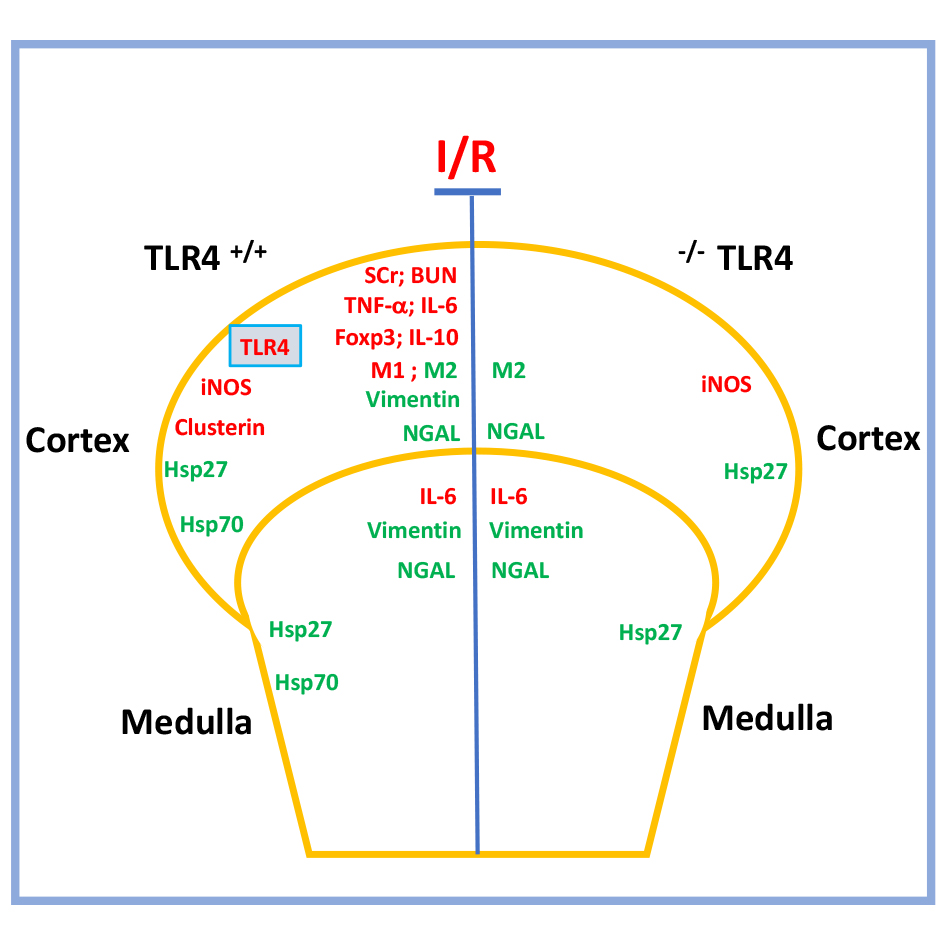
Considering the above-described aspect, we use a murine model of IRI in wild-type (WT) and Knockout (KO) for TLR4 to explore new potential molecular targets for TLR4 during IRI in the cortex and medulla kidney independently. We have found that the absence of TLR4 during IRI prevented the impaired renal blood depuration function but not the tubular damage. The TLR4-KO reduced the upregulation of proinflammatory (IL-6 and TNF-α), anti-inflammatory (Foxp3 and IL-10) factors, and M1 (CD38 and Fpr2) macrophage markers, especially in the cortex. Interestingly, IL-6 was upregulated in the cortex and medulla independently of TLR4. In addition, the M2 macrophage markers (Erg-2 and c-Myc) were upregulated by IRI only in the cortex and it was independent of TLR4. Vimentin was upregulated in the cortex and medulla by IRI in TLR4-WT and TLR4-KO inhibited vimentin upregulation only in the renal cortex. Besides, iNOS and clusterin were upregulated cortex by IRI in WT and the absence of TRL4 inhibits clusterin. Finally, Hsp27 and Hsp70 were upregulated by IRI in both groups of animals and the absence of TRL4 inhibits Hsp70. In conclusion, TLR4 participates in ischemia and reperfusion through pro-inflammatory and anti-inflammatory responses inducing impaired kidney function (SCr and BUN). However, the upregulation of M2 macrophage markers (cortex), iNOS (cortex), IL-6 (medulla), mesenchymal markers (medulla), Hsp27 (cortex and medulla) by IRI were induced by IRI independent of TLR4.
Materials and Methods
Animals
Adult males, 8-12 weeks old (20–22 g), from C57BL/6 mice, and C57BL/10ScNJ mice (20–22 g) with a targeted disruption of the Toll-like receptor 4 genes (TLR4-KO) were obtained from Jackson Laboratory (Bar Harbor, ME). Genotypic analyses were performed according to technical data suggested by Jackson Laboratory. Mice were housed in a controlled environment, provided with standard rodent chow, water, and maintained at the Universidad de los Andes Animal Care Facility. All experimental procedures were by accordance with institutional and international standards for the humane care and use of laboratory animals (Animal Welfare Assurance Publication A5427-01, Office for Protection from Research Risks, Division of Animal Welfare. The National Institutes of Health). All procedures were approved by the Committee on the Ethics of Animal Experiments of the Universidad de los Andes, Chile.
Ischemia-reperfusion (I/R)
The animals were anesthetized with 25mg/kg i.p. ketamine /15mg/kg i.p. xylazine and maintained on a 37°C blanket during the surgical procedure. Both kidneys were exposed by a flank incision and the renal pedicle was occluded for 30 min with a non-traumatic vascular clamp (cat N° 18055-02 Fine Science Tools). Renal blood flow was re-established (reperfusion stage) by clamp removal and both incisions were sutured. Sham animals did not undergo renal pedicle occlusion [23, 26]. Reperfusion was 48 hrs. Following protocols, mice were euthanized with CO2, kidneys were dissected and processed for Western blotting, qRT-PCR, histology, and biochemical analyses.
Assessment of Renal Function
The serum creatinine (SCr) and blood urea nitrogen (BUN) levels were measured by an automatic analyzer (Mindray analyzer).
Real-Time PCR
The protocol was conducted as it was described previously [23, 27]. In brief, the total RNA was isolated using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s directions. Extracted RNA was quantified at 260-nm in a NanoDrop Spectrophotometer (NanoDrop Technologies) and the integrity of the RNA was assessed by agarose gel electrophoresis. cDNA was prepared from total RNA (1μg) using a reverse transcription system (random hexamers, Improm II Reverse Transcriptase System from Promega). Then, PCR was performed in duplicate for each experiment (HotStart Taq DNA polymerase from Qiagen or BRILLIANT III ULTRA-FAST SYBR GREEN QPCR (Stratagene). The primers used are listed in Table 1. Amplicons were detected for Real-Time Fluorescence Detection (Rotor-Gene Q, Qiagen). Relative mRNA abundance was calculated using Ct values and normalized to the relative abundance of each transcript.
Histochemical analysis and tissue damage determination
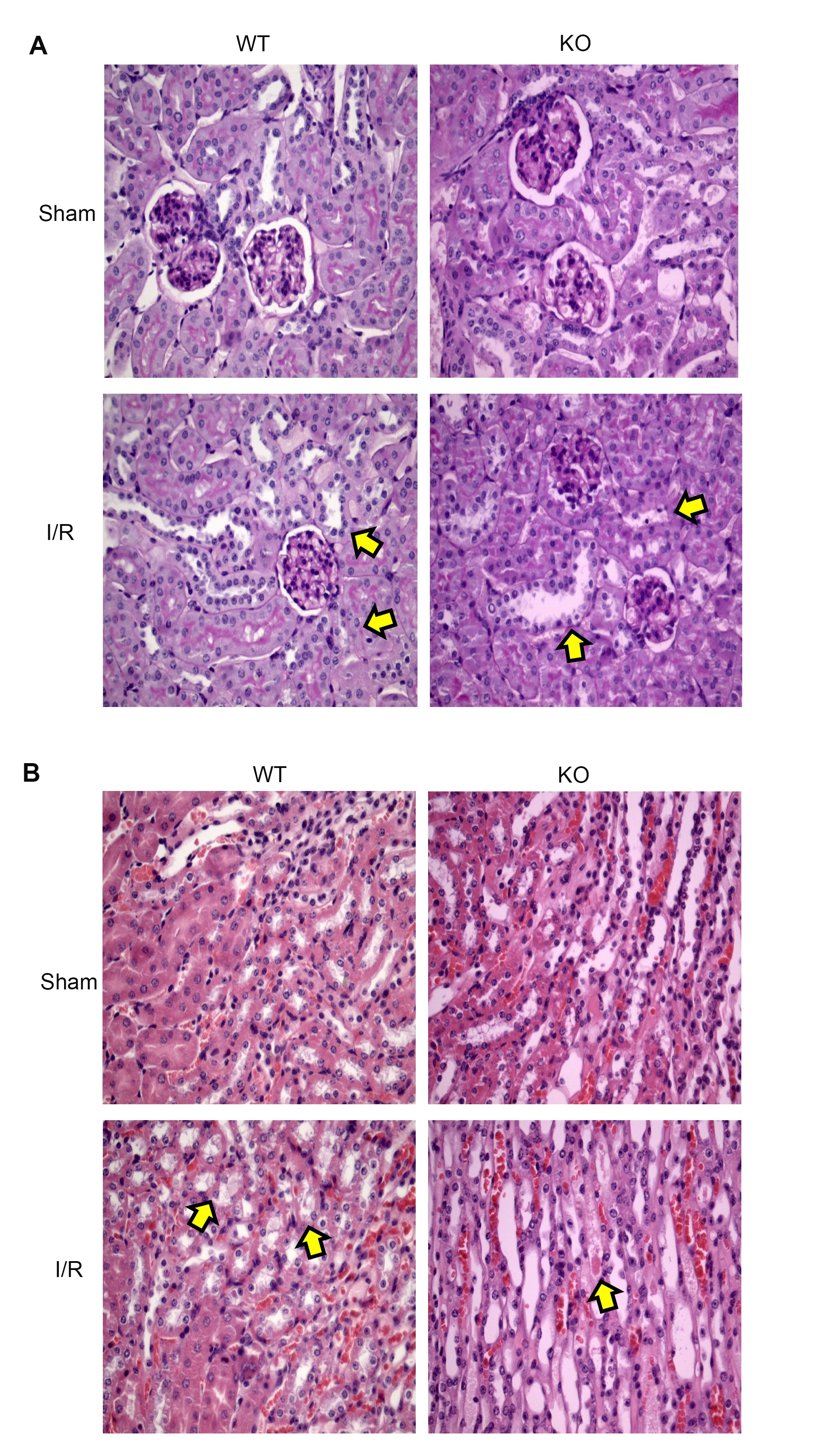
The kidneys were fixed in 10% buffered formalin, embedded in paraffin, sectioned, dewaxed, rehydrated, rinsed in water, and stained with hematoxylin and eosin and periodic acid Schiff (PAS). The morphologic analysis was carried out in a blinded manner as detailed previously [23, 28]. The cortex and medulla were evaluated for epithelial necrosis, loss of brush border, tubular dilation, and tubular congestion among other kidney alterations observed in response to AKI.
Western blot assay
Western blot was realized as was previously published with some modifications [27–29]. Briefly, the renal cortex and medulla were dissected and homogenized with an Ultra-Turrax homogenizer in ice-cooled 10 mM Tris·HCl buffer at pH 7.4, supplemented with 1 mM EDTA, 1 mM EGTA, 0.25 M sucrose, 1% vol/vol Triton X-100, and a protease inhibitor cocktail (Complete Mini, Roche Applied Science). Tissue homogenates were subject to the next steps. Homogenized was centrifugated to 3,000 rpm by 10 min (4°C). Next, the tissue was sonicated for 30min on ice, high-speed vortex by 1min, and centrifugated again at 3,000 rpm by 10min (4°C). The supernatant produced was centrifugated to 5,000rpm by 5 min (4°C). Finally, the supernatant was centrifugated to 5,000rpm by 5 min and centrifugated to 14,000rpm by 5 min (4°C). Total proteins in supernatants were measured using the BCA Protein Assay Kit (ThermoFisher Scientific), and samples were stored at -80°C. The antibodies used were anti-NGAL (Abcam), anti-Hsp27 (cell signaling), anti-TLR4 (Santa Cruz, Biotechnology), and anti-β-actin (Sigma). The antibody anti-Hsp70 was provided by Dr Gabriele Multhoff. Secondary antibodies were anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor-750 (Thermo Scientific). Intensities of the resulting bands were quantified using Odyssey equipment and Image Studio Lite software (version 5.25; Li-Cor).
Statistical analysis
Differences between groups were analyzed using the non-parametric Mann-Whitney U test or post-hoc Tukey test using GraphPad Prism Software. The level of significance was set at p < 0.05. Data are presented as the mean ± standard error of the mean (SEM).
Results
Characterization of TLR4 expression
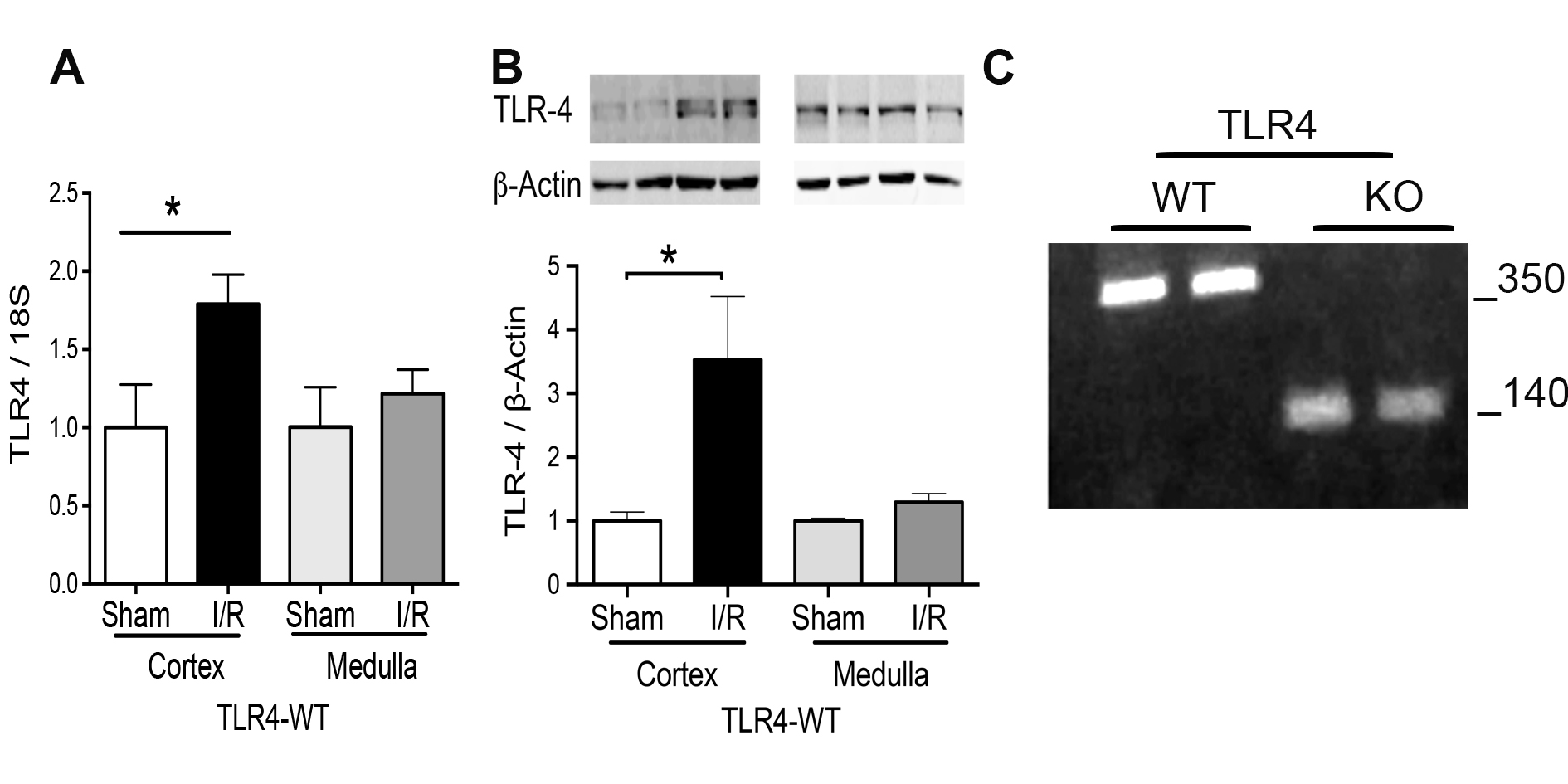
We used 30 min of ischemia and 48 hours of the reperfusion according to our and other publications [23, 25, 30, 31]. The cortex and medulla were analyzed separately and we found in this timing that TLR4 (mRNA and protein) was significantly induced by I/R in the cortex but not in the medulla of TLR4-WT animals (Fig. 1A and 1B). In addition, the TLR4-WT and TLR4-KO condition was verified by genotypic analyses in the kidney of each enrolled animal according to technical data suggested by Jackson Laboratory (Fig. 1B).
Effect of TLR4 on kidney injury during renal ischemia/reperfusion injury (IRI)
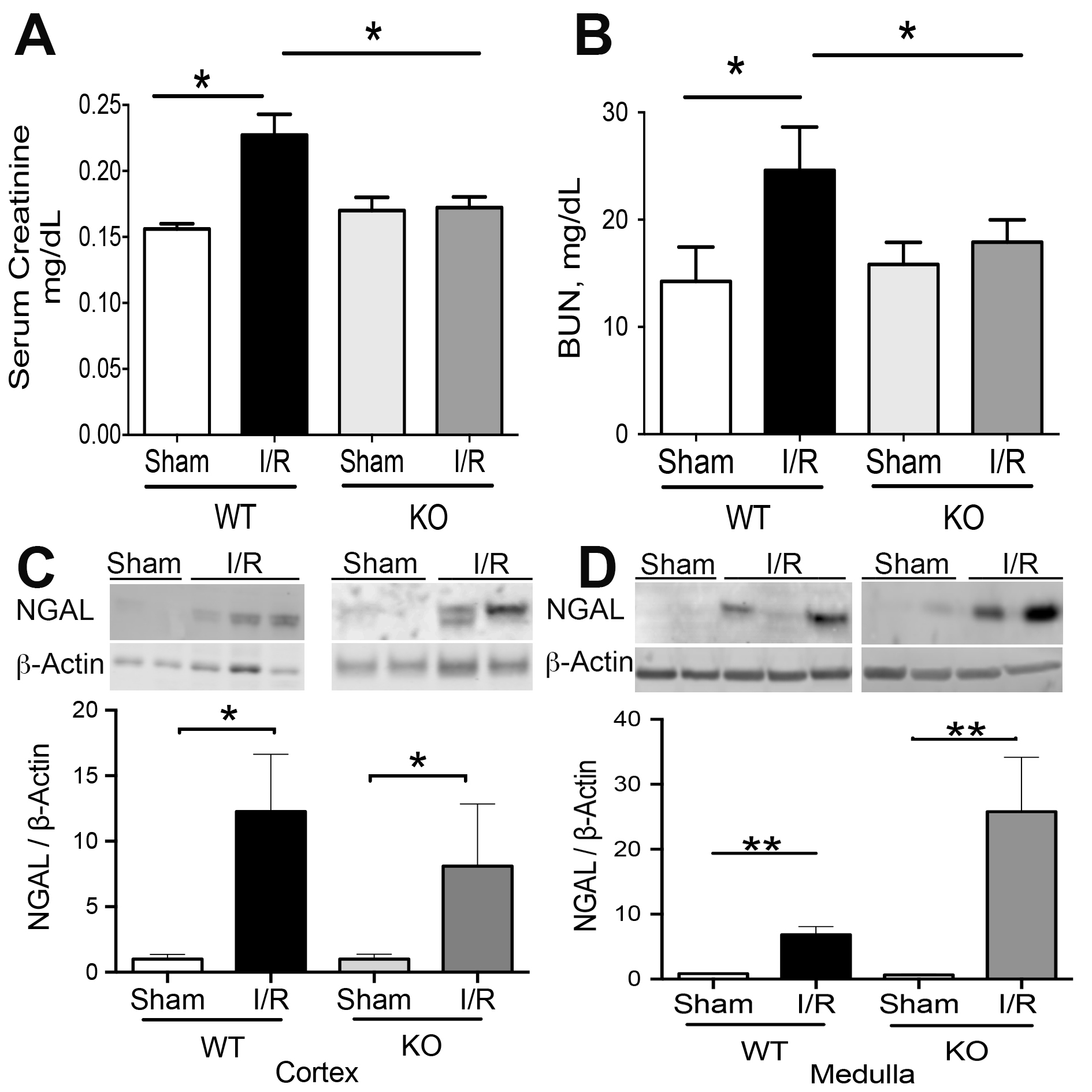
The kidney injury was studied by measuring the levels of NGAL, serum creatinine (SCr), blood urea nitrogen (BUN), and histological observation of Acute Tubular Necrosis (ATN) in TLR4-WT and TLR4-KO mice. The SCr and BUN (Fig. 2A-B) levels increased by renal I/R in TLR4-WT but not in the TLR4-KO group, confirming previously published results [8, 11]. However, the NGAL protein abundance increased significantly by IRI in the cortex and medulla belonging to TLR4-WT and TLR4-KO mice (Fig. 2C-D). Moreover, the ATN sign was observed in both types of animals, characterized by epithelial cell disruption, brush border loss, and tubular congestion (see arrowheads in Fig. 3A). The PAS-stained sections showed the same results (Fig. 3B). No changes were observed in the kidney tissues from both controls (sham) belonging to TLR4-WT or TLR4-KO mice. Altogether, these results suggest that blocking the TLR4 expression has a partial effect on renal protection during renal IRI.
TLR4 on the inflammatory response during renal IRI
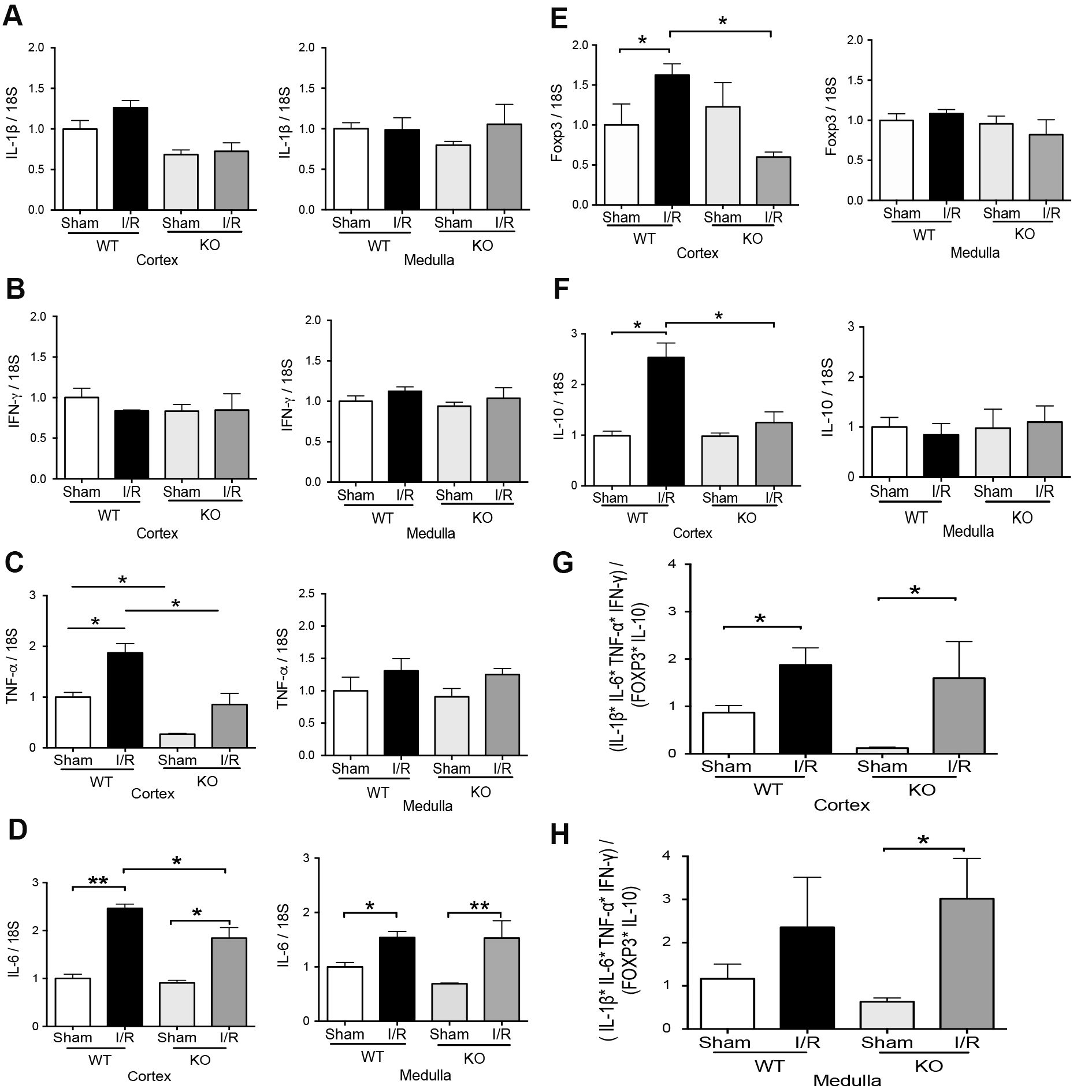
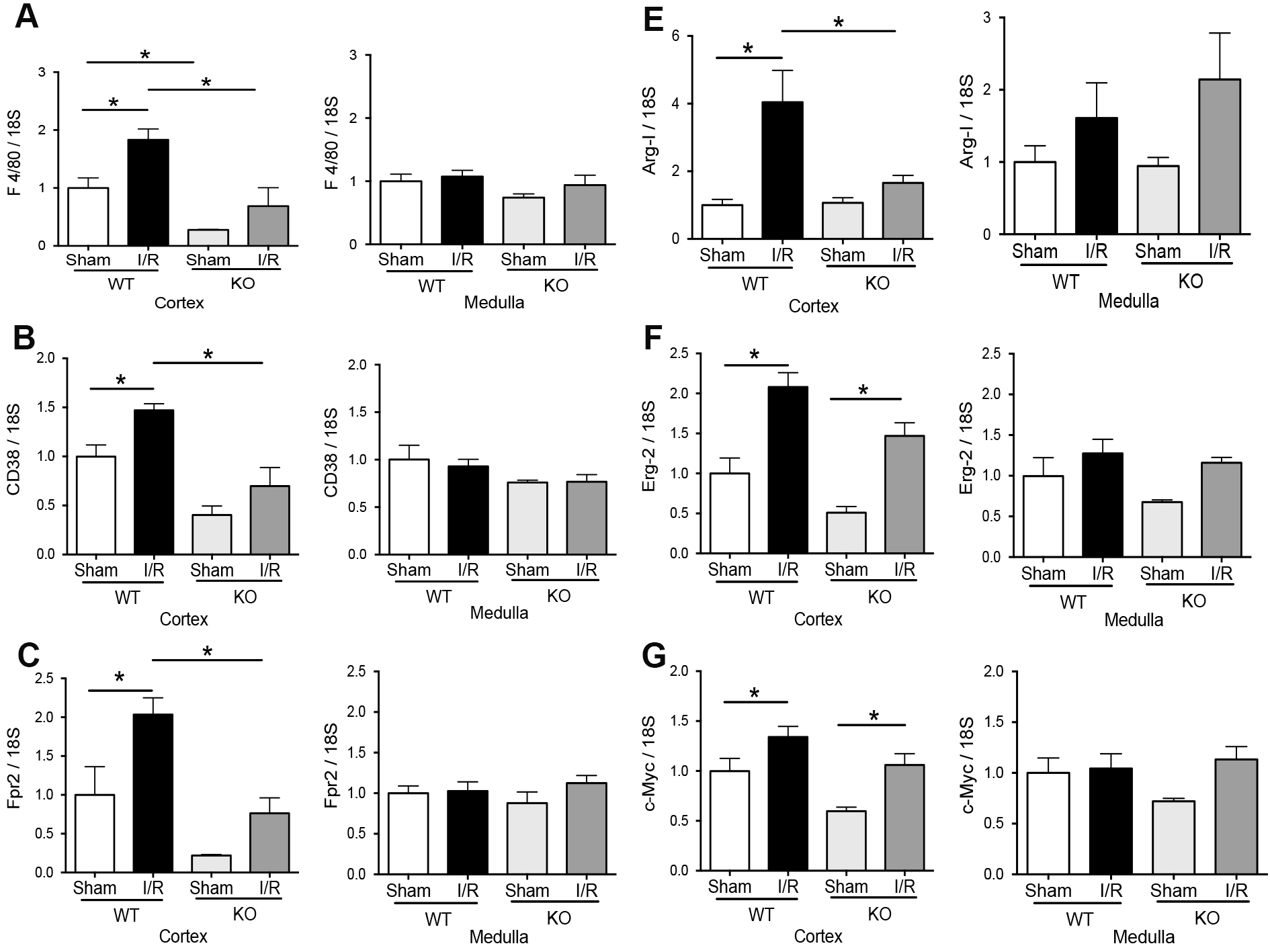
We investigated the expression of pro-inflammatory (TNF-α, IFN-γ, IL-1β, and IL-6) and regulatory (IL-10 and Foxp3) factors in TLR4-WT and TLR4-KO mice at 48 hours of reperfusion. The IL-1β and IFN-γ mRNA did not change by IRI after 48hrs of reperfusion in the two kidney sections (cortex or medulla) of both kinds of animals (Fig. 4A-B). Conversely, IL-6 mRNA increased significantly in the cortex and medulla from the TLR4-WT and TLR4-KO mice by renal IRI compared with the respectively sham group. Interestingly, the IL-6 had a partial but significant reduction in the mRNA expression during IRI in TLR4-KO than in TLR4-WT. In addition, TNF-α mRNA was significantly induced in the cortex but not in the medulla by IRI in TLR4-WT. The TLR4-KO animals did not increase the TNF-α mRNA (Fig. 4C-D) by IRI. On the other hand, in the kidney cortex, the Foxp3 and IL-10 mRNA were upregulated by renal IRI in TLR4-WT mice but not in TLR4-KO animals (Fig. 4E-F). In addition, in the kidney medulla, Foxp3 and IL-10 mRNA did not experiment changes by IRI in TLR4-WT or TLR4-KO animals. We established a relationship between IL-1β, IFN-γ, IL-6, TNF-α, Foxp3, and IL-10 mRNA levels in each animal studied by a simple equation ([IL-1β] x [IL-6] x [IFN-γ] x [TNF-α] / [Fox3] x [IL-10]) as an inflammatory index. The results showed that there are no differences in the inflammatory index induced by IRI in both kinds of animals (Fig. 4G-H). Interestingly, the macrophage markers showed that M0 (F4/80), M1(CD38 and Fpr2), and M2 (Arg-1, Egr-2, and c-Myc) were upregulated by IRI in the cortex but not in the medulla in TLR4-WT animals. Remarkably, the TLR4-KO inhibited the M1 but not the M2 macrophage polarization (Fig. 5A-G) in the cortex kidney. The overall results suggested that during renal IRI, TLR4 participates in the molecular signaling of both proinflammatory and anti-inflammatory factors in the cortex kidney. Interestingly, the medullary expression of TLR4 was not upregulated by IRI and it was associated with the less inflammatory signal in our model of IRI.
Effect of TLR4 on mesenchymal markers by renal IRI
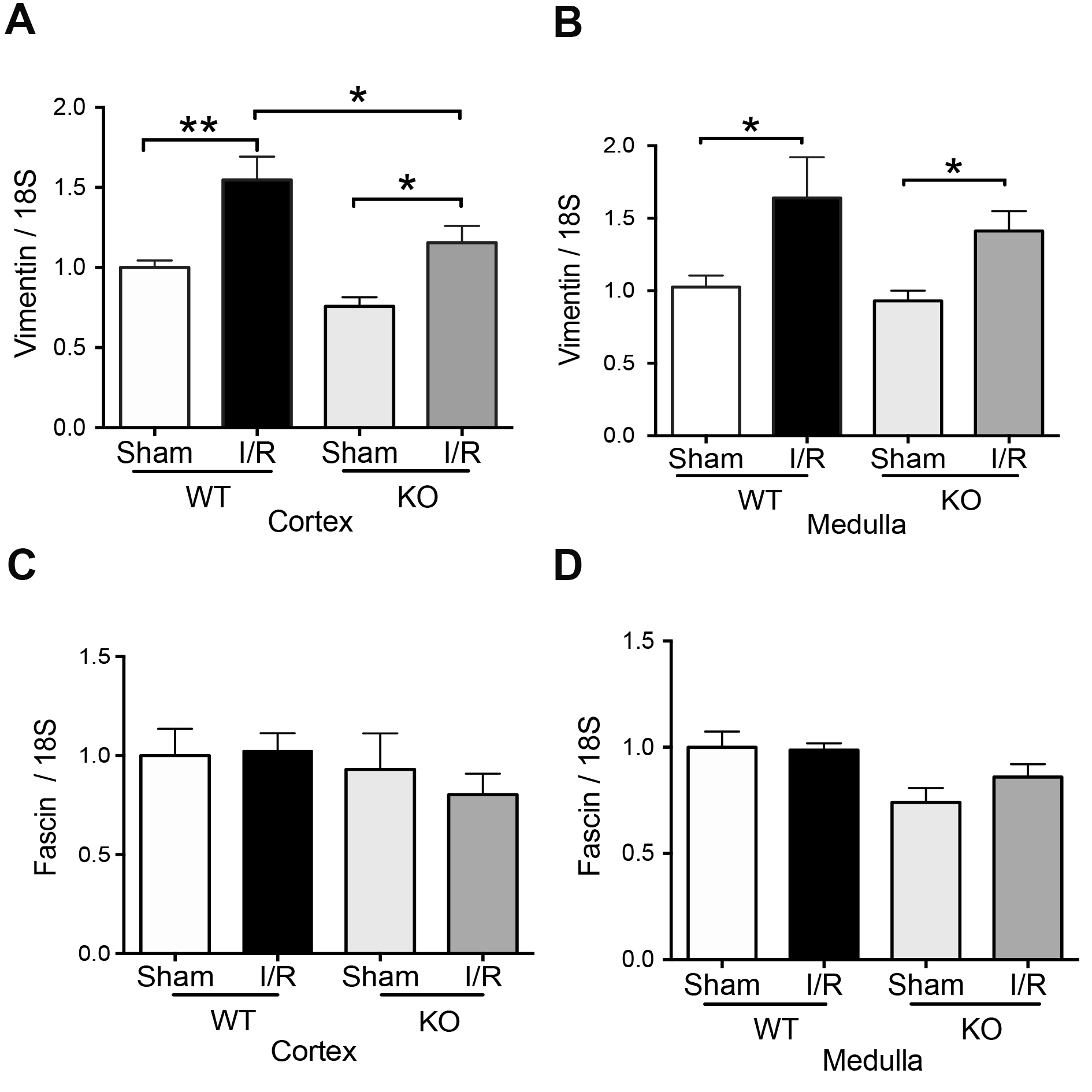
Previously was described that a panel of mesenchymal markers (vimentin and fascin) are overexpressed in human kidney allograft, experimenting with acute tubular necrosis [13] and renal ischemia and reperfusion injury in mice [25]. We found increased mRNA levels of Vimentin in the cortex kidney (Fig. 6A) belonging to TLR4-WT mice during IRI. Notably, in the cortex of TLR4-KO animals, IRI did not reach the levels of Vimentin mRNA observed in TLR4-WT but still was significantly higher than in TLR4-KO sham animals (Fig. 6A). In contrast, in the medulla, the Vimentin was upregulated by IRI in both TLR4-WT and TLR4-KO mice compared with respectively sham animals (Fig. 6B). The fascin mRNA did not change in TLR4-WT or TLR4-KO by IRI (Fig. 6C-D). Considering this information, we propose that the mesenchymal transition observed during IRI is blunt only in the cortex in the TLR4-KO condition.
TLR4 effect on iNOS and clusterin expression by renal IRI
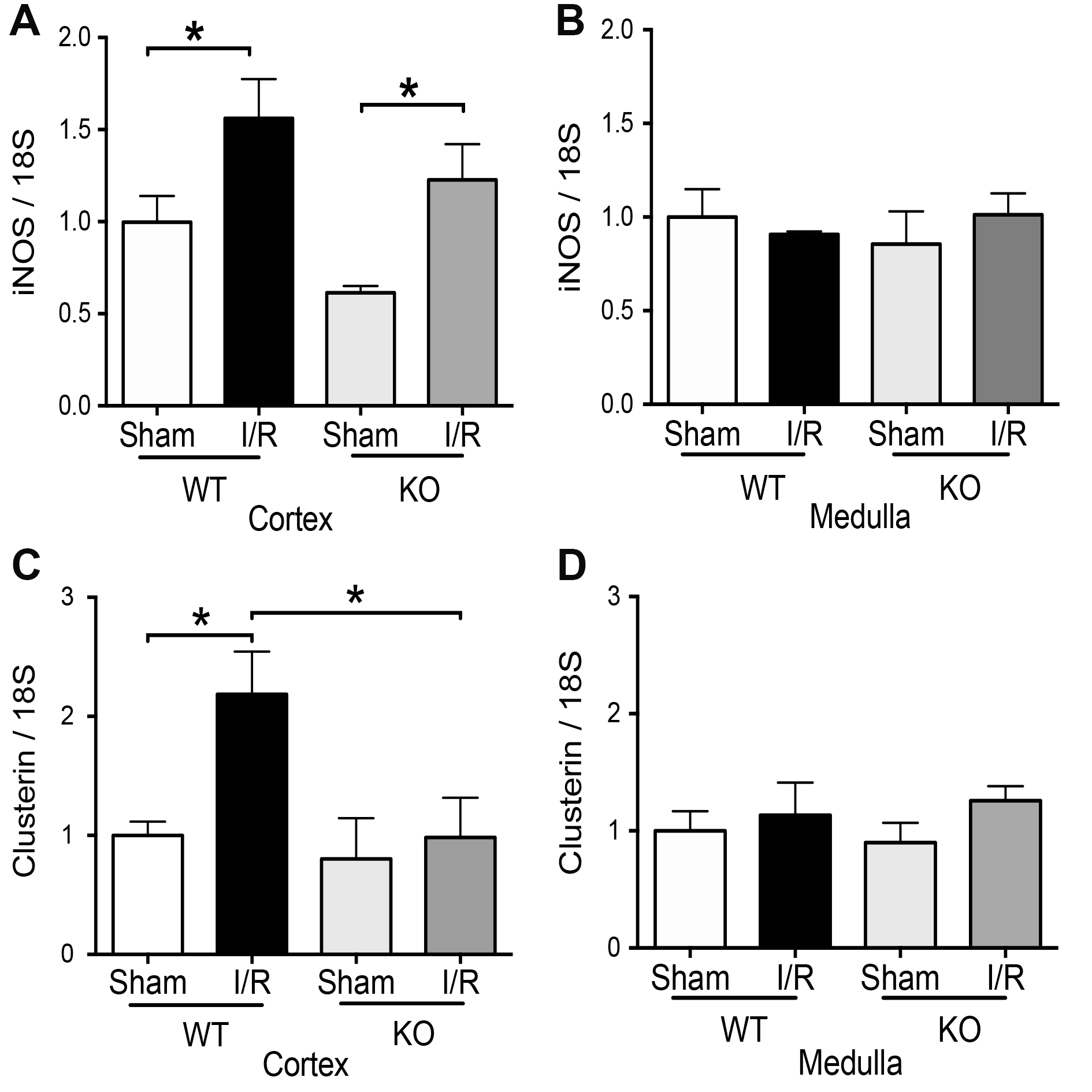
The inducible form of nitric oxide synthase (iNOS) increases by IRI and has detrimental effects on kidneys by IRI [23, 24]. We previously published that iNOS and clusterin increase in the murine model of IRI [23] and the pharmacological inhibition of iNOS (L-NIL), before IRI, improves renal function and prevent clusterin upregulation after 48hrs of reperfusion [23]. In addition, we recently described that aminoguanidine (antioxidant) prevented the upregulation of clusterin and iNOS induced by IRI [30]. Here, we observed upregulation of iNOS mRNA by IRI in the cortex of TLR4-WT and TLR4-KO mice (Fig. 7A-B), suggesting that TLR4 is not the only upstream regulator of iNOS expression during IRI. In addition, here we observed that clusterin mRNA was also significantly increased by IRI in the cortex from TLR4-WT but not in TLR4-KO mice in the kidney cortex (Fig. 7C). No effect in clusterin mRNA was observed in the kidney medulla in both types of mice by IRI (Fig. 7D).
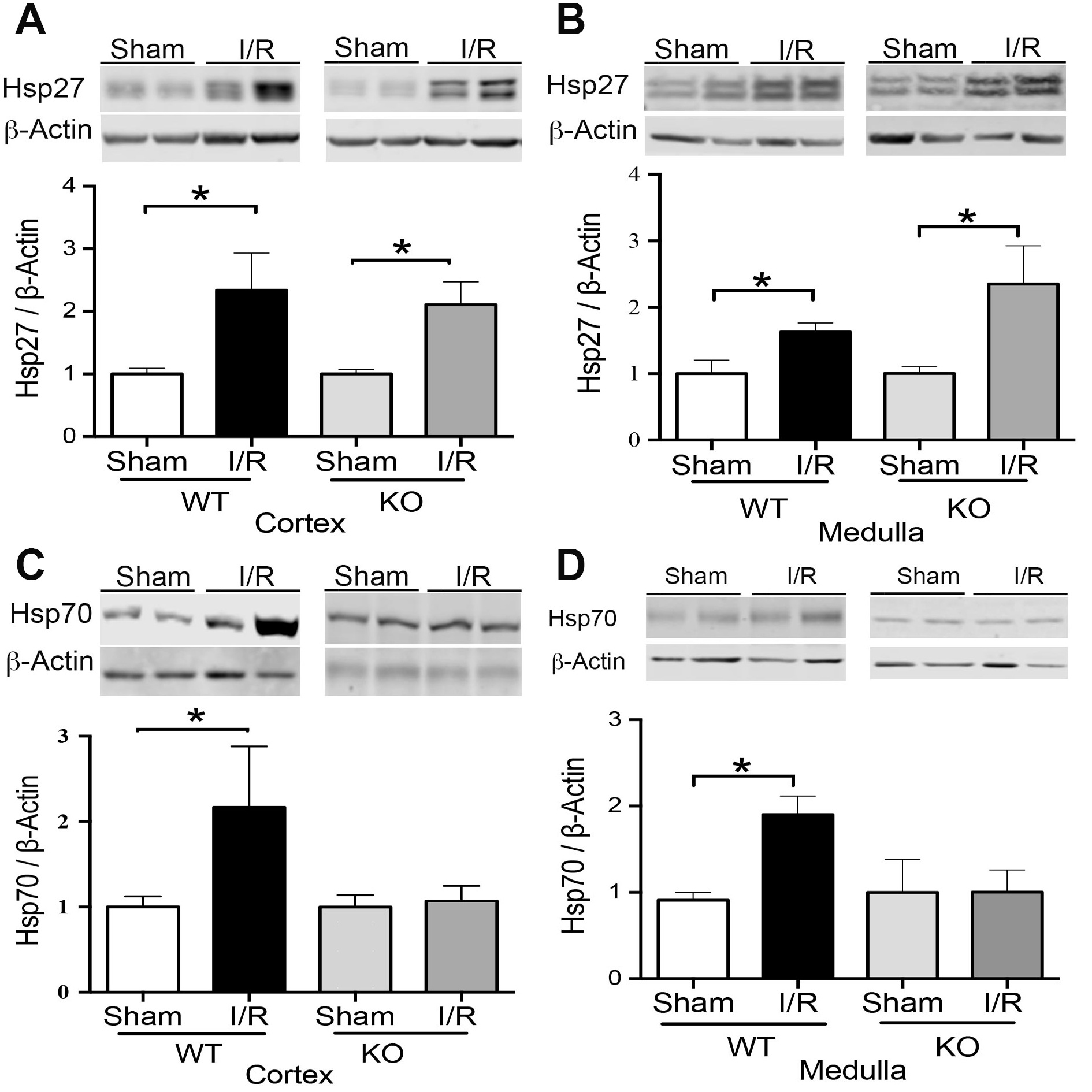
TLR4 effect on Hsp27 and Hsp70 expression by renal IRI
Finally, we studied the effect of TLR4 on the expression of heat shock proteins 27 (Hsp27) and 70 (Hsp70). We noted that the Hsp27 protein expression increased significantly in TLR4-WT and TLR4-KO mice by IRI, both in the cortex and medulla compared with sham animals (Fig. 8A-B), suggesting that TLR4 is not an upstream pathway of Hsp27. On the other hand, Hsp70 increased by IRI in the kidney after 48hrs of reperfusion in TLR4-WT animals. Noteworthy, the TRL4-KO inhibited the Hsp70 upregulation by IRI (Fig. 8C-D). This data suggests that TLR4 might be an upstream regulator of Hsp70 expression during IRI.
The absence of TLR4 did not prevent other signs of tubular damage (NGAL and ATN), inflammation (IL-6), and oxidative stress (Hsp27 and iNOS)
Neutrophil gelatinase-associated lipocalin (NGAL) is synthesized by epithelial cells of the tubule as a primary response to IRI [48]. The NGAL mRNA is synthesized primarily in the thick ascending limb and collecting duct and is not expressed in proximal tubules [49]. Here we found NGAL protein upregulation by IRI in the cortex and medulla of TLR4-WT animals, and the absence of TLR4 did not prevent this upregulation, suggesting that tubular damage produced by IRI is independent of TLR4 expression. The histopathological analysis confirmed those results (Fig. 3). On the other hand, after 48hrs of reperfusion, we did not see any changes in IL-1β or IFN-γ mRNA by IRI in both groups of animals, but the IL-6 mRNA was upregulated independently of TLR4 in the cortex and medulla. Remarkably, Foxp3 and IL-10 mRNA increased by IRI only in the cortex but not in the medulla of TLR4-WT. The IRI-upregulation of TNF-α in the cortex by IRI in TLR4-WT was reduced in TLR4-KO animals. Remarkably, the absence of TLR4 completely inhibited the Foxp3 and IL-10 mRNA upregulation by IRI. According to a previous publication, we found that TLR4-KO produces a partial but significant reduction in IL-6 mRNA than TLR4-WT [7, 50]. It noteworthy, previously was published that IL-10 mRNA was higher in TLR4-KO than TLR4-WT during IRI [11] after 24hrs of reperfusion. However, we observed that IL-10 mRNA decreased when TLR4 was absent in the kidney cortex after 48hrs of reperfusion.
We previously described that TLR4 and iNOS increase by IRI. Interestingly, we observed that an iNOS inhibitor (l-NIL) prevented the TLR4 upregulation by IRI [23]. Here, we found that iNOS was upregulated by IRI independently of TLR4, suggesting that other effectors more than TLR4 participate in iNOS upregulation by IRI. On the other side, Heat shock protein 27 (Hsp27) has been described to have a role as a chaperone, antioxidant, inhibitor of apoptosis, and works as an actin cytoskeletal remodeling [16]. Interestingly, kidney-specific expression of Hsp27 through lentiviral produces a renal protective role of this chaperone during IRI [15], reducing apoptosis, necrosis, and pro-inflammatory cytokines. These mice also demonstrated better F-actin preservation. We found Hsp27 upregulation by IRI in the cortex and medulla of TLR4 WT and KO animals. Thus, we suggest that Hsp27 was induced by IRI independently of TLR4.
TLR4 is a necessary player in the activation of the innate immune response. TLR4 recognizes damage-associated molecular patterns and activates secondary immune responses. In addition, TLR4 might also be activated in podocytes, mesangial, tubular epithelial, and endothelial cells in the kidneys and have a renoprotective function through IL-22 production. We found that the TLR4 deficiency during IRI improved the renal blood depuration function (reduced SCr and BUN). In addition, TLR4-KO inhibited in the cortex the polarization of the M1 macrophages (CD38 and Fpr2), mesenchymal markers (Vimentin and Fascin), clusterin, Hsp70, Foxp3, and IL-10 upregulation, increasing the evidence of TLR4 is involved into increase the response to the injury produced by ischemia-reperfusion. Because in the renal medulla, the elimination of TLR4 did not prevent the upregulation of vimentin during IRI, we suggest that TLR4 has a differential effect in the cortex than medulla.
Conclusion
In summary, TLR4 participates in ischemia and reperfusion through pro-inflammatory and anti-inflammatory responses inducing impaired kidney function (SCr and BUN). However, the IRI-upregulation of M2 macrophage markers (cortex), iNOS (cortex), IL-6 (medulla), mesenchymal markers (medulla), and Hsp27 (cortex and medulla) were independent of TLR4. Therefore, the TLR4 inactivation before IRI prevented the loss of renal function, inactivation of inflammation response, avoiding M1, and preserving the M2 macrophage polarization in the renal cortex.
The authors thank Dr. Luis Contreras for his generous help in the detection of pathological kidney in mice.
Author Contributions
Yeimi Herrera-Luna: Investigation (equal). Mauricio Lozano: Investigation (equal). Consuelo Pasten: Investigation (equal); Writing-review & editing (equal). Gabriele Multhoff: Resources (equal). Carlos E. Irarrázabal: Conceptualization (equal); Funding acquisition (equal); Writing-original draft (equal); Writing-review & editing (equal).
Funding
The present study was financially supported through grants from the FONDECYT-1151157 and FAI-Universidad de los Andes.
The authors have no conflicts of interest to declare.
| 1 Vázquez-Carballo C, Guerrero-Hue M, García-Caballero C, Rayego-Mateos S, Opazo-Ríos L, Morgado-Pascual JL, Herencia-Bellido C, Vallejo-Mudarra M, Cortegano I, Gaspar ML, de Andrés B, Egido J, Moreno JA: Toll-Like Receptors in Acute Kidney Injury. Int J Mol Sci 2021;22:816. https://doi.org/10.3390/ijms22020816 |
||||
| 2 Jha AK, Gairola S, Kundu S, Doye P, Syed AM, Ram C, Murty US, Naidu VGM, Sahu BD: Toll-like receptor 4: An attractive therapeutic target for acute kidney injury. Life Sci 2021;271:119155. https://doi.org/10.1016/j.lfs.2021.119155 |
||||
| 3 Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M: Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One 2015;10:e0145342. https://doi.org/10.1371/journal.pone.0145342 |
||||
| 4 Chen H, Zhang RQ, Wei XG, Ren XM, Gao XQ: Mechanism of TLR-4/NF-κB pathway in myocardial ischemia reperfusion injury of mouse. Asian Pac J Trop Med 2016;9:503-507- https://doi.org/10.1016/j.apjtm.2016.03.021 |
||||
| 5 Chen J, Yang C, Xu X, Yang Y, Xu B: The effect of focal cerebral ischemia-reperfusion injury on TLR4 and NF-κB signaling pathway. Exp Ther Med 2018;15:897-903. https://doi.org/10.3892/etm.2017.5463 |
||||
| 6 Zanotti G, Casiraghi M, Abano JB, Tatreau JR, Sevala M, Berlin H, Smyth S, Funkhouser WK, Burridge K, Randell SH, Egan TM: Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol Physiol 2009;297:L52-63. https://doi.org/10.1152/ajplung.90406.2008 |
||||
| 7 Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ: TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 2007;117:2847-2859. https://doi.org/10.1172/JCI31008 |
||||
| 8 Pulskens WP, Teske GJ, Butter LM, Roelofs JJ, van der Poll T, Florquin S, Leemans JC: Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 2008;3:e3596. https://doi.org/10.1371/journal.pone.0003596 |
||||
| 9 El-Achkar TM, Huang X, Plotkin Z, Sandoval RM, Rhodes GJ, Dagher PC: Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am J Physiol Ren Physiol 2006;290:1034-1043. https://doi.org/10.1152/ajprenal.00414.2005 |
||||
| 10 Zhao H, Perez JS, Lu K, George AJT, Ma D: Role of toll-like receptor-4 in renal graft ischemia-reperfusion injury. Am J Physiol - Ren Physiol 2014;306:F801-811. https://doi.org/10.1152/ajprenal.00469.2013 |
||||
| 11 Chen CB, Liu LS, Zhou J, Wang XP, Han M, Jiao XY, He XS, Yuan XP: Up-Regulation of HMGB1 Exacerbates Renal Ischemia-Reperfusion Injury by Stimulating Inflammatory and Immune Responses through the TLR4 Signaling Pathway in Mice. Cell Physiol Biochem 2017;41:2447-2460. https://doi.org/10.1159/000475914 |
||||
| 12 Kulkarni OP, Hartter I, Mulay SR, Hagemann J, Darisipudi MN, Kumar Vr S, Romoli S, Thomasova D, Ryu M, Kobold S, Anders HJ: Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. J Am Soc Nephrol 2014;25:978-989. https://doi.org/10.1681/ASN.2013050528 |
||||
| 13 Xu-Dubois YC, Ahmadpoor P, Brocheriou I, Louis K, Arzouk Snanoudj N, Rouvier P, Taupin JL, Corchia A, Galichon P, Barrou B, Giraud S, Hauet T, Jouanneau C, Rodenas A, Placier S, Niasse A, Ouchelouche S, Naimi BY, Akil E, Hertig A, Buob D, Rondeau E: Microvasculature partial endothelial mesenchymal transition in early posttransplant biopsy with acute tubular necrosis identifies poor recovery renal allografts. Am J Transplant 2020;20:2400-2412. https://doi.org/10.1111/ajt.15847 |
||||
| 14 Guo Q, Du X, Zhao Y, Zhang D, Yue L, Wang Z: Ischemic postconditioning prevents renal ischemia reperfusion injury through the induction of heat shock proteins in rats. Mol Med Rep 2014;10:2875-2881. https://doi.org/10.3892/mmr.2014.2641 |
||||
| 15 Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D'Agati VD, Lee HT: Selective renal overexpression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 2010;299:F347-F358. https://doi.org/10.1152/ajprenal.00194.2010 |
||||
| 16 Vidyasagar A, Wilson NA, Djamali A: Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenes Tissue Repair 2012;5:1-7. https://doi.org/10.1186/1755-1536-5-7 |
||||
| 17 Kim MG, Jung Cho E, Won Lee J, Sook Ko Y, Young Lee H, Jo SK, Cho WY, Kim HK: The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4+ CD25+ Foxp3 + regulatory T cells in ischemia/reperfusion-induced acute kidney injury. Kidney Int 2014;85:62-71. https://doi.org/10.1038/ki.2013.277 |
||||
| 18 Gandolfo MT, Jang HR, Bagnasco SM, Ko GJ, Agreda P, Satpute SR, Crow MT, King LS, Rabb H: Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int 2009;76:717-729. https://doi.org/10.1038/ki.2009.259 |
||||
| 19 Alnasser HA, Guan Q, Zhang F, Gleave ME, Nguan CY, Du C: Requirement of clusterin expression for prosurvival autophagy in hypoxic kidney tubular epithelial cells. Am J Physiol Renal Physiol 2016;310:F160-F173. https://doi.org/10.1152/ajprenal.00304.2015 |
||||
| 20 Witzgall R, Brown D, Schwarz C, Bonventre JV: Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 1994;93:2175-2188. https://doi.org/10.1172/JCI117214 |
||||
| 21 Guo J, Guan Q, Liu X, Wang H, Gleave ME, Nguan CY, Du C: Relationship of clusterin with renal inflammation and fibrosis after the recovery phase of ischemia-reperfusion injury. BMC Nephrol 2016;17:133. https://doi.org/10.1186/s12882-016-0348-x |
||||
| 22 Nguan CYC, Guan Q, Gleave ME, Du C: Promotion of cell proliferation by clusterin in the renal tissue repair phase after ischemia-reperfusion injury. Am J Physiol Ren Physiol 2014;306:724-733. https://doi.org/10.1152/ajprenal.00410.2013 |
||||
| 23 Pasten C, Alvarado C, Rocco J, Contreras L, Aracena P, Liberona J, Suazo C, Michea L, Irarrázabal CE: l-NIL prevents the ischemia and reperfusion injury involving TLR-4, GST, clusterin, and NFAT-5 in mice. Am J Physiol Renal Physiol 2019;316:F624-F634. https://doi.org/10.1152/ajprenal.00398.2018 |
||||
| 24 Mark LA, Robinson A V., Schulak JA: Inhibition of nitric oxide synthase reduces renal ischemia/reperfusion injury. J Surg Res 2005;129:236-41. https://doi.org/10.1016/j.jss.2005.06.019 |
||||
| 25 Pasten C, Herrera-Luna Y, Lozano M, Rocco J, Alvarado C, Liberona J, Michea L, Irarrázabal CE: Glutathione S-Transferase and Clusterin, New Players in the Ischemic Preconditioning Renal Protection in a Murine Model of Ischemia and Reperfusion. Cell Physiol Biochem 2021;55:635-650. https://doi.org/10.33594/000000442 |
||||
| 26 Wei Q, Dong Z: Mouse model of ischemic acute kidney injury: technical notes and tricks. Am J Physiol Renal Physiol 2012;303:F1487-F1494. https://doi.org/10.1152/ajprenal.00352.2012 |
||||
| 27 Serman Y, Fuentealba RA, Pasten C, Rocco J, Ko BCB, Carrión F, Irarrázabal CE: Emerging new role of NFAT5 in inducible nitric oxide synthase in response to hypoxia in mouse embryonic fibroblast cells. Am J Physiol Cell Physiol 2019;317:C31-C38. https://doi.org/10.1152/ajpcell.00054.2019 |
||||
| 28 Figueroa H, Alvarado C, Cifuentes J, Lozano M, Rocco J, Cabezas C, Illanes SE, Eixarch E, Hernández-Andrade E, Gratacós E, Irarrazabal CE: Oxidative damage and nitric oxide synthase induction by surgical uteroplacental circulation restriction in the rabbit fetal heart. Prenat Diagn 2017;37:453-459. https://doi.org/10.1002/pd.5031 |
||||
| 29 Ugarte F, Irarrazabal C, Oh J, Dettmar A, Ceballos ML, Rojo A, Ibacache MJ, Suazo C, Lozano M, Delgado I, Cavada G, Azocar M, Delucchi A, Cano F: Impaired phosphorylation of JAK2-STAT5b signaling in fibroblasts from uremic children. Pediatr Nephrol 2016;31:965-974. https://doi.org/10.1007/s00467-015-3289-x |
||||
| 30 Pasten C, Lozano M, Rocco J, Carrión F, Alvarado C, Liberona J, Michea L, Irarrázabal CE: Aminoguanidine Prevents the Oxidative Stress, Inhibiting Elements of Inflammation, Endothelial Activation, Mesenchymal Markers, and Confers a Renoprotective Effect in Renal Ischemia and Reperfusion Injury. Antioxidants 2021;10:1724. https://doi.org/10.3390/antiox10111724 |
||||
| 31 Pasten C, Lozano M, Méndez GP, Irarrázabal CE: 1400W Prevents Renal Injury in the Renal Cortex But Not in the Medulla in a Murine Model of Ischemia and Reperfusion Injury. Cell Physiol Biochem 2022 Oct 19;56:573-586. https://doi.org/10.33594/000000577 |
||||
| 32 Brown HJ, Lock HR, Wolfs TGAM, Buurman WA, Sacks SH, Robson MG: Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol 2007;18:1732-1399. https://doi.org/10.1681/ASN.2006060634 |
||||
| 33 Chen J, Hartono JR, John R, Bennett M, Zhou XJ, Wang Y, Wu Q, Winterberg PD, Nagami GT, Lu CY: Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int 2011;80:504-515. https://doi.org/10.1038/ki.2011.140 |
||||
| 34 Wolfs TG, Buurman WA, van Schadewijk A, de Vries B, Daemen MA, Hiemstra PS, van 't Veer C: In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol 2002;168:1286-1293. https://doi.org/10.4049/jimmunol.168.3.1286 |
||||
| 35 Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S: Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest 2005;115:2894-2903. https://doi.org/10.1172/JCI22832 |
||||
| 36 Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB: TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol 2007;178:6252-6258. https://doi.org/10.4049/jimmunol.178.10.6252 |
||||
| 37 Paulus P, Rupprecht K, Baer P, Obermüller N, Penzkofer D, Reissig C, Scheller B, Holfeld J, Zacharowski K, Dimmeler S, Schlammes J, Urbschat A: The early activation of toll-like receptor (TLR)-3 initiates kidney injury after ischemia and reperfusion. PLoS One 2014;9:e94366. https://doi.org/10.1371/journal.pone.0094366 |
||||
| 38 Alvarez S, Suazo C, Boltansky A, Ursu M, Carvajal D, Innocenti G, Vukusich A, Hurtado M, Villanueva S, Carreño JE, Rogelio A, Irarrazabal CE: Urinary exosomes as a source of kidney dysfunction biomarker in renal transplantation. Transplant Proc 2013;45:3719-3723. https://doi.org/10.1016/j.transproceed.2013.08.079 |
||||
| 39 Zhang FR, Tao LH, Shen ZY, Lv Z, Xu LY, Li EM: Fascin expression in human embryonic, fetal, and normal adult tissue. J Histochem Cytochem 2008;56:193-199. https://doi.org/10.1369/jhc.7A7353.2007 |
||||
| 40 Yamakita Y, Matsumura F, Lipscomb MW, Chou PC, Werlen G, Burkhardt JK, Yamashiro S: Fascin1 promotes cell migration of mature dendritic cells. J Immunol 2011;186:2850-2859. https://doi.org/10.4049/jimmunol.1001667 |
||||
| 41 Shim YJ, Tae YK, Kang BH, Park JS, Jeon SY, Min BH: Toll-like receptor 4 signaling is required for clusterin-induced tumor necrosis factor-α secretion in macrophage. Biochem Biophys Res Commun 2017;482:1407-1412. https://doi.org/10.1016/j.bbrc.2016.12.049 |
||||
| 42 Zhou J, Chen X, Gilvary DL, Tejera MM, Eksioglu EA, Wei S, Djeu JY: HMGB1 induction of clusterin creates a chemoresistant niche in human prostate tumor cells. Sci Rep 2015;5:15085. https://doi.org/10.1038/srep15085 |
||||
| 43 Wang Z, Gall JM, Bonegio RG, Havasi A, Hunt CR, Sherman MY, Schwartz JH, Borkan SC: Induction of heat shock protein 70 inhibits ischemic renal injury. Kidney Int 2011;79:861-870. https://doi.org/10.1038/ki.2010.527 |
||||
| 44 Suzuki S, Maruyama S, Sato W, Morita Y, Sato F, Miki Y, Kato S, Katsuno M, Sobue G, Yuzawa Y, Matsuo S: Geranylgeranylacetone ameliorates ischemic acute renal failure via induction of Hsp70. Kidney Int 2005;67:2210-2220. https://doi.org/10.1111/j.1523-1755.2005.00326.x |
||||
| 45 Jheng HF, Tsai PJ, Chuang YL, Shen YT, Tai TA, Chen WC, Chou CK, Ho LC, Tang MJ, Lai KT, Sung JM, Tsai YS: Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech 2015;8:1311-1321. https://doi.org/10.1242/dmm.019398 |
||||
| 46 Stangl S, Gehrmann M, Riegger J, Kuhs K, Riederer I, Sievert W, Hube K, Mocikat R, Dressel R, Kremmer E, Pockley AG, Friedrich L, Vigh L, Skerra A, Multhoff G: Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc Natl Acad Sci U S A 2011;108:733-738. https://doi.org/10.1073/pnas.1016065108 |
||||
| 47 Specht HM, Ahrens N, Blankenstein C, Duell T, Fietkau R, Gaipl US, Günther C, Gunther S, Habl G, Hautmann H, Hautmann M, Huber RM, Molls M, Offner R, Rödel C, Rödel F, Schütz M, Combs SE, Multhoff G: Heat Shock Protein 70 (Hsp70) Peptide Activated Natural Killer (NK) Cells for the Treatment of Patients with Non-Small Cell Lung Cancer (NSCLC) after Radiochemotherapy (RCTx) - From Preclinical Studies to a Clinical Phase II Trial. Front Immunol 2015;6:162. https://doi.org/10.3389/fimmu.2015.00162 |
||||
| 48 Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, Viltard M, Yu W, Forster CS, Gong G, Liu Y, Kulkarni R, Mori K, Kalandadze A, Ratner AJ, Devarajan P, Landry DW, D'Agati V, Lin CS, Barasch J: The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 2011;17:216-222. https://doi.org/10.1038/nm.2290 |
||||
| 49 Desanti De Oliveira B, Xu K, Shen TH, Callahan M, Kiryluk K, D'Agati VD, Tatonetti NP, Barasch J, Devarajan P: Molecular nephrology: types of acute tubular injury. Nat Rev Nephrol 2019;15:599-612. https://doi.org/10.1038/s41581-019-0184-x |
||||
| 50 Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, Chadban SJ. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol 2010;21:1878-1890. https://doi.org/10.1681/ASN.2009101048 |
||||