Epistemology of the Origin of Cancer II: Fibroblasts Are the First Cells to Undergo Neoplastic Transformation
Keywords
Abstract
Background/Aims:
Many questions in cancer biology remain unanswered. Perhaps the most important issues remaining to be addressed focus on the molecular basis of carcinogenesis. Today’s cancer focus lies on genetics and gene expression, which is unlikely to explain the true cause of most cancers or lead to a cure.Methods:
Earlier, we provided a plausible mechanism for this process, specifically, that most cancers develop in response to pathogenic stimuli that induce chronic inflammation, fibrosis, and remodeling of the cellular microenvironment. Collectively, these changes generate a precancerous niche (PCN) in which fibrosis and remodeling are ongoing secondary to persistent inflammation, followed by the deployment of a chronic stress escape strategy (CSES). If the CSES is unsuccessful, the cell undergoes a normal cell to cancer cell transformation (NCCT).Results:
Here, we highlight the critical role of fibroblasts as the first cells to undergo neoplastic transformation to a cancerous phenotype which is based on several critical findings. First, persistent disruption of homeostatic crosstalk increases lysyl oxidase activity and lysine oxidation which leads to increased collagen stiffness and decreased elasticity. If unresolved, chronic tissue stress will lead to an escape strategy that involves the recruitment of fibroblasts and fibrocytes from the bone marrow as well as cells undergoing an epithelial-mesenchymal transition (EMT). This yields a heterogeneous pool of cells that express both epithelial and mesenchymal markers and that will ultimately differentiate into cancer-associated fibroblasts (CAFs). Finally, CAFs undergo a mesenchymal-epithelial transition (MET) and express epithelial markers that facilitate their integration into the target tissue.Conclusion:
Here, we review the published findings that led us to this conclusion which is the most plausible answer to this critical question.Introduction
Among the major issues in cancer medicine remaining to be addressed is the question of why the increased patient survival is measured in weeks and months but not in years. One reason for this conundrum is that cancer biology remains incompletely understood. We have only a limited appreciation of important issues related to cancer diversity [1, 2], heterogeneity [3–12], and resistance to therapy [13–16], as well as those leading to failed pretreatment strategies [17], and sensitivity testing [18]. Thus far, only a small proportion of cancer patients have truly benefitted from existing therapies [19]. While many believe that the discovery of new cancer-specific genes and epitopes will lead to promising drug candidates for “personalized” therapy [20], real-world data suggest that the gains from these efforts typically are smaller than anticipated [21, 22]. Hence, many basic concepts in cancer biology still need to be considered (Supplement Part 1, Cancer basics).
Since the acievements of COVID mRNA vaccines, such vaccines against cancer are seen as the game changers for the future [23, 24], although we know that the stock market is driven by psyschology [25]. Clearly, cause-based approaches, for example, extensive use of the vaccine designed to combat human papillomaviruses (HPV), has made a significant difference in the clinical outcomes of cancers associated with this pathogen. Note that, in this case, these viruses are the known cause of most cervical and oropharyngeal cancers. With appropriate use of the antiviral vaccine, these cancers can be virtually eliminated [26, 27].
There is a need for a more precise and in-depth understanding of the essential causes of and mechanisms underlying carcinogenesis. Toward this end, the first important question that needs to be answered is how specific signaling and crosstalk pathways contribute to the development of cancer. We addressed this issue in our earlier paper entitled “Epistemology of the Origin of Cancer: a new cancer paradigm” [28] as well as in several other publications [29–43]. In the first of these aforementioned papers [28], we reviewed findings that suggested that most cancers originate after a defined sequence of events, beginning with (1) a pathogenic stimulus, followed by (2) chronic inflammation that leads to (3) fibrosis and associated changes in the cellular microenvironment. If this state persists, (4) a pre-cancerous niche (PCN) develops, which triggers the deployment of (5) a chronic stress escape strategy (CSES). If this condition fails to resolve, (6) normal cells may transition to cancer cells (NCCCT) occurs. A great deal of literature supporting this paradigm has been published in recent years (Supplement Part 2, Recent evidence (since 2019)).
Also remaining is the question of which cell type is the first to undergo normal cell-to-cancer cell transition. In this paper, we provide a plausible, albeit complex, set of findings that fibroblasts are the critical cells that undergo NCCCT in response to pathogenic and inflammatory stimuli in most human cancers.
Epithelial ultrastructure
Because ~90% of all cancers are comprised of epithelial cells (Supplement Part 1, Cancer basics), the literature on this subject focuses on this cell type and its physiologic ultrastructure. One of the critical structural elements of the epithelium is the basement membrane (also known as the basal membrane, or lamina basalis), which can be found at the interface between cells and the underlying stroma that includes several sub-laminal layers [44–46]. The basement membrane not only promotes tissue stability, it also plays an active role in the complex internal and external crosstalk and signaling pathways within a living organism. The basement membrane includes three layers known as the lamina densa, the lamina rara interna, and the lamina fibroreticularis, which can be delineated and differentiated by electron microscopy. A simplified schematic of this structure is presented in Figure 1. While earlier reports mentioned a lamina rara interna (lamina lucida), this structure has since been identified as a fixation artifact [47].
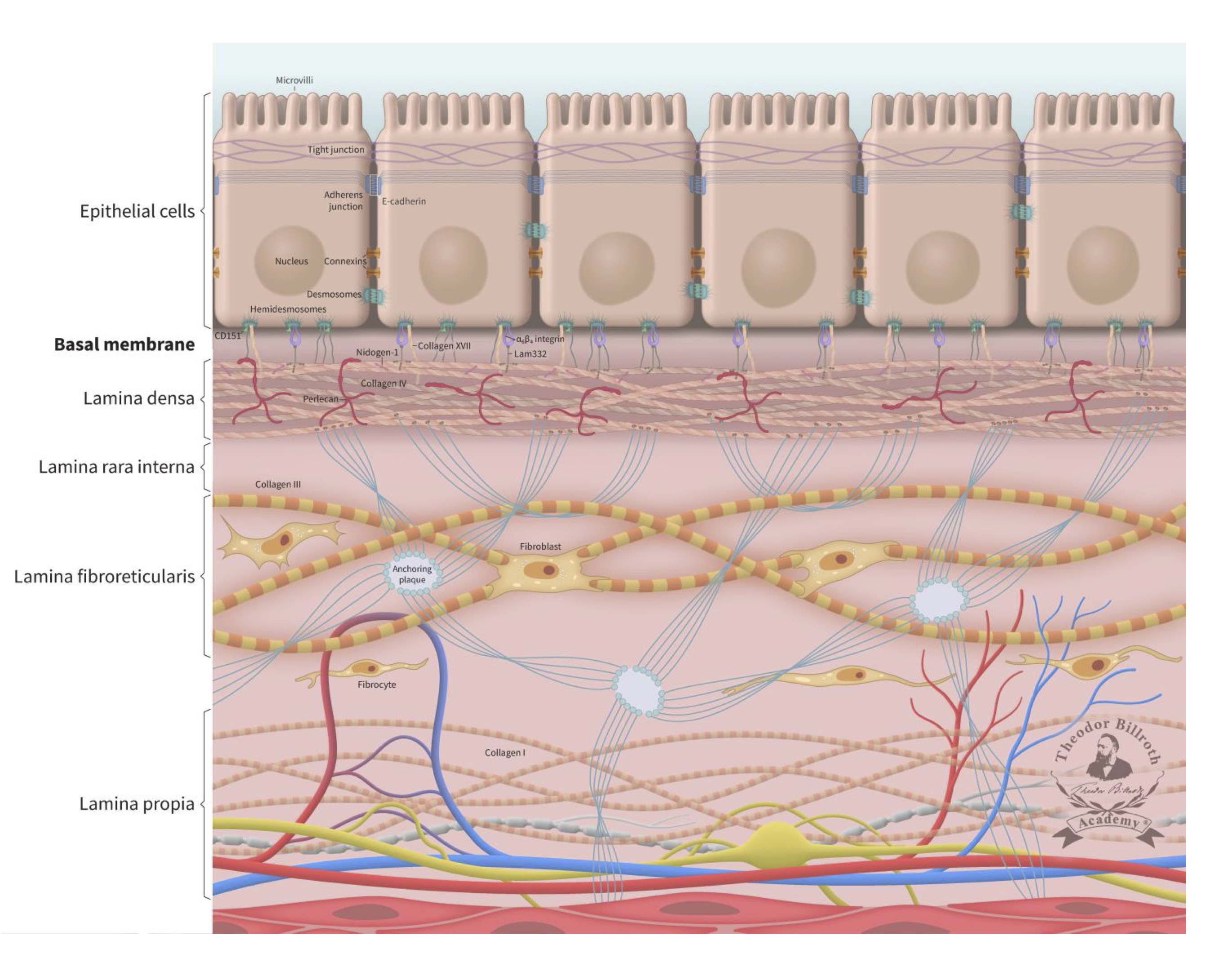
Fig. 1: Simplified ultrastructure of the epithelium and the basal membrane (basement membrane). The cell-cell junctions with its communication, occluding and anchoring junctions relevant for cell-cell communications were reviewed in detail earlier together with a schematic drawing of junctions between epithelial cells [29]. This schematic drawing is enlarged with E-cadherin and the underlying basal membrane (basement membrane) and the lamina propria. The lamina densa immediately beneath the basal membrane has an average thickness of 50 nm (ranging from 20 to 300 nm). The thickness of the lamina fibroreticularis is typically 200 to 500 nm. The basal membrane consist out of the lamina densa, lamina rara interna and lamina fibroreticularis. The lamina densa consist out of Collagen IV, Perlecan, Nidogen-1 (entactin), laminin 332 (Lam332), laminin311, and laminin 511. The interconnection to the epithelium is organized via Collagen XIII, Collagen VII, cluster of differentiation 151 (CD151), 64 integrin, 31 integrin, laminin 332, and laminin 511. To improve the overview of the simplified schematic structure, some components are not shown (Collagen XIII, 31 integrin, laminin 311, laminin 511 and other components). The lamina fibroreticularis consists of Collagen III (reticular fibers), and anquoring plaques (Collagen IV) and Collagen VIII and includes fibroblasts. Their inactive form, fibrocytes, are below located in the lamina propria within the network of Collagen I, arteriols, venols, and lymphatic drainage. The epithelial layer is connected to the sublayer space of the lamina densa by keratin intermediate filaments, 64-integrins, 31 integrins, bullous pemphigoid antigen 1 (BPAG1, also known as dytonin, DST), plectin, collagen-type XVII (Col17), and cluster of differentiation 151 (CD151) of the hemidesmosomes. These cells also express surface receptor proteins (i.e., CD44 and laminin receptors). The connection to the lamina densa mainly consists of anchoring filaments, which are composed of laminin 332 and the extracellular COL17A1, 64 integrins, CD151, laminin 311, and laminin 511. The lamina densa contains collagen-type IV (Col4), laminin 332, laminin 311, laminin 511, nidogen-1 (entactin), and perlecan (also known as heparan sulfate proteoglycan 2, HSPG2). The lamina fibroreticularis consists of collagen-type III (Col3, i.e., reticular fibers), collagen-type IV (Col4, i.e., anchoring plaques), and collagen-type VII (Col7, i.e., anchoring fibrils). Fibroblasts play a significant role in maintaining the ultrastructure and stability of these layers. Additional relevant historical remarks and information on fibroblasts and fibroblast biology are provided in Supplement Part 3, Fibroblasts: historical consideration.
Fibroblasts and stiffness
Fibroblasts are ubiquitous cells derived from the embryonic mesenchyme that are found predominantly outside the lamina densa. Fibroblasts are capable of producing and secreting all extracellular matrix (ECM) components required by adipocytes as well as macrophages, mast cells, and other leukocytes [43, 48].
Fibroblast responses to chemical carcinogens
Fibroblast cultures generated from normal mammalian tissue that were treated with the carcinogen, 20-methylchloranthrene, for four months to as long as four years converted into epithelial-like cells and became laterally adherent, depending on the duration of treatment [49]. When these cells were injected into mice, tumors arose at the injection site. Interestingly, tumors also arose in mice injected with fibroblasts that had not been exposed to the carcinogen, a finding that was attributed to the unique and specific properties of the fibroblasts themselves [50, 51]. After numerous passages in vitro over the course of three years, the incidence of carcinoma dropped from 68% to 1%. Because no other explanation could be found at the time, this observation was attributed to the acquisition of one or more functional mutations. Today, these findings would be recognized as a primary mesenchymal-epithelial cell transition (MET) that was followed three years later by an epithelial-mesenchymal cell transition (EMT). While we now have a clear mechanistic understanding of this observation, we can reflect on the fact that many unexplained findings were, and continue to be, attributed to mutational events [30].
The same group then explored two different clones of the same fibroblast strain and identified one that was highly aggressive (carcinoma incidence of 97%) and another that was more benign (carcinoma incidence of 1%); however, these results were observed only in mice that had first been irradiated; this was attributed to differences in immunity [52]. Furthermore, this group reported that mouse cell cultures that included stromal cells with epithelial glandular architecture underwent a cancerous transformation after 1.5 years and 35 transplantation generations in vitro and were capable of generating mammary carcinomas [53]. Similarly, fibroblasts infected with Rous Sarcoma Virus (RSV) became spindle-shaped (fibroblast-like) or amoeboid phagocytes; after re-implantation, the fibroblast-like cell tumors grew and persisted, while the amoeboid cells always gave rise to tumors [54]. Taken together, these findings reveal how migrating fibroblasts promoted continuous cancer growth in vitro and likewise induced an immunosuppressive PCN, as this phenomenon is currently described [55]. Transformed cancer cells exhibit high glycolytic activity, which, intriguingly, was reported to be even greater than the enzymatic activities of whole cells or extracts calculated on a per-cell basis [56]. Of note, increased glycolytic activity was detected in rapidly-growing cultures long before cancerous transformation and, interestingly, virus-infected and uninfected cells exhibited the same morphologic changes related to loss of contact inhibition [57] including a decrease in the number of anchoring junctions [29].
Spontaneous transformation of cells with morphological features typical of both cancerous and non-cancerous cells was accompanied by an increased nuclear:cytoplasmic ratio, cytoplasmic basophilia, and a change in the size and shape of cells and their nuclei even before the transformation process first occurred. Furthermore, results from multiple experiments revealed that transition to a cancer cell phenotype was inhibited in the presence of fetal bovine serum [58]. Of note, the first tumor cell generations exhibited prominent cytological changes, including changes in cell shape and reduced spreading; this was followed by more random migration patterns. Cancer cell transition was also associated with morphological changes that included a decrease in the projected area of cytoplasmic lamellae, the nucleus, and the dry mass of the lamellar cytoplasm.
In 1960, Sir John Gurdon performed experiments in frogs that proved cells were capable of changing after undergoing differentiation. Among the findings, he reported that these cells could revert not only to a more primitive state but also to one with higher developmental potential [59] and differentiated cells retained this ability [60, 61].
Interestingly, scientists working at that time did not perceive the transition of fibroblasts into neoplastic epithelial cells as a general model of carcinogenesis, as discussed in reference [62]. Epithelial-like cells revealed different agglutination properties and expression patterns [63]; which may explain in part why fibroblasts were not considered as initial cancer precursor cells despite many lines of converging data. Even later on, this transition stage was not recognized as a phenomenon of critical importance. As noted by Hentzer et al. [64], “…unidentifiable cells were characterized by dense zones of the cell membrane and an adjacent extracellular basal lamina-like material…”. In this context, we recall that cardiac fibroblasts originate from epithelial cells of the proepicardium in a developing embryo via a process known as EMT. Likewise, valvular fibroblasts arise via endothelial-mesenchymal transition (EndMT) of cells in the endocardium [65].
Conversion of fibroblasts to stem cells
In 1981, Evans and Kauffman [66] identified, isolated, and cultured embryonic stem cells from mice. Somewhat later, Takahashi et al. [67, 68] altered four specific genes and successfully converted skin-derived fibroblasts into pluripotent stem (iPS) cells. Similarly, Gordon et al. [69], reported that cortisone-treated cultured fibroblasts could undergo conversion into adipocytes. In other experiments, brown fat precursors were identified as adipocyte stem cells, although cell differentiation in these experiments remained incomplete [70]. Furthermore, various cell types were available in these experiments, including “…fibroblasts, adipocytes, and adipose precursor cells before determination (adipoblasts) and after determination (preadipocytes)…” [71]. Collectively, these studies led to the introduction of the concept of stem cells [72].
Fibrocytes are fibroblasts in their inactive form and found predominantly within the lamina propria in a region beyond the lamina densa. Epithelial migration takes place during embryologic development and fibroblasts are first detected under the basal membrane and they migrate during maturation to the alveolar wall [73]. Importantly, fibroblasts can develop from epithelial cell precursors during the process of tissue fibrosis [74]. Additional fibroblasts can be recruited from the deeper layers, which also are a source of circulating fibrocytes [75]. Furthermore, fibroblasts from bone marrow can be recruited to fibrotic lesions [76] and importantly, also to the PCN by the neoplastic cells themselves [38].
Chronic inflammation secondary to tumor growth factor beta (TGF-β1)-mediated pathways, including those that lead to both Smad and non-Smad JNK/AP-1 signaling results in increased collagen production by fibroblasts with lysyl oxidase (LOX) activity [28] (FIGURE 2). Several previous studies reviewed the origin and nature of LOX and its isoforms which are triggered by the complex cascade of chronic inflammation to generate fibrosis and the PCN [28, 38]. Copper and lysyl tyrosyl quinone are both critical co-factors that facilitate the LOX-mediated conversion of lysine residues to α-aminoadipidic-δ-semialdehydes (allysines) in collagen and elastin precursors.
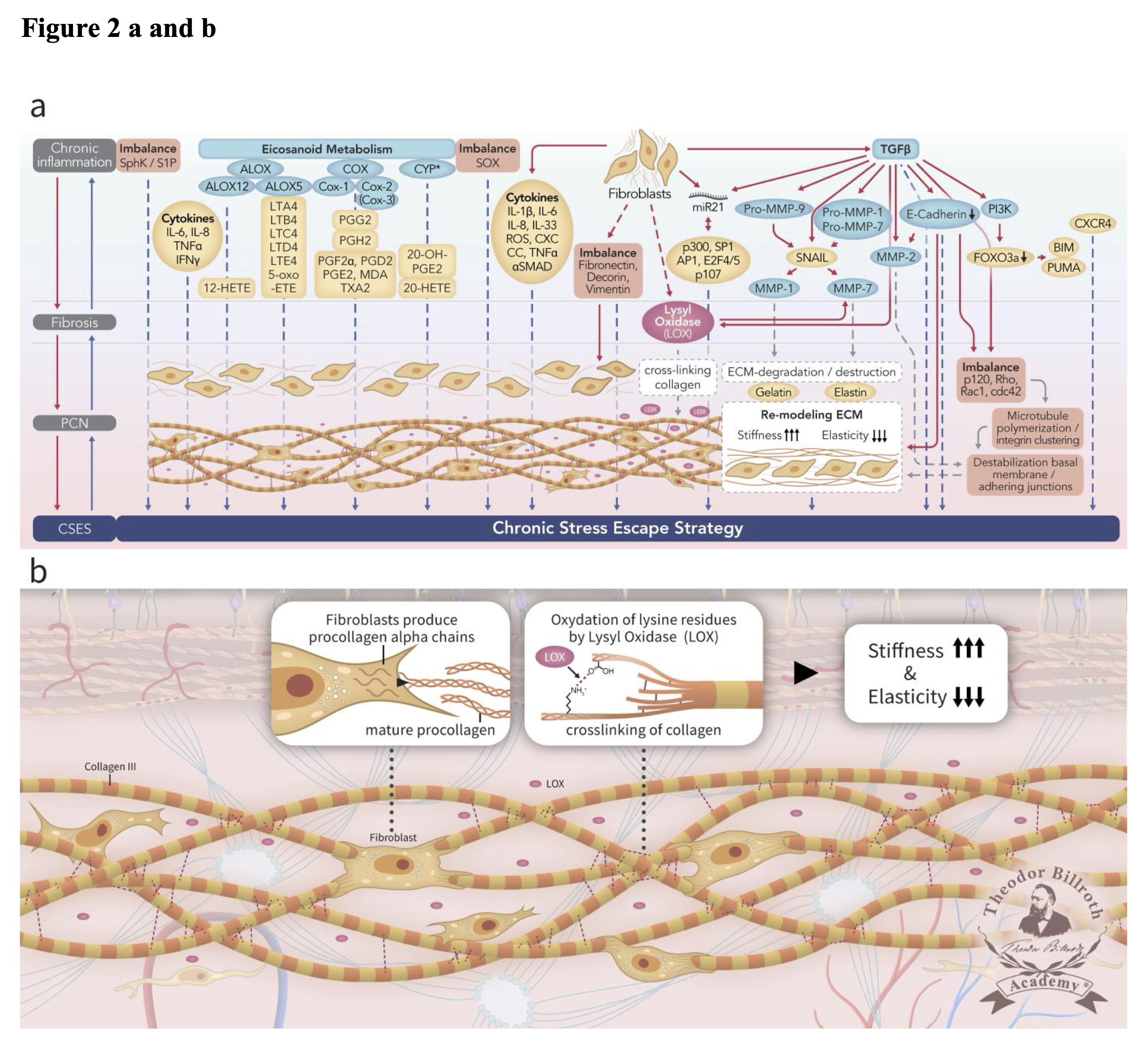
Fig. 2: Simplified scheme representing ongoing disruption of homeostatic crosstalk originally discussed in the paradigm presented in our earlier publication [28] in the upper drawn (Fig. 2a) and depicting the lamina fibroreticularis layer and its precancerous niche (PCN) in the lower drawn as a cutout (Fig. 2b). As shown in the upper drawn (Fig. 2a), a pathogenic stimulus induces chronic inflammation which eventually leads to fibrosis with remodeling of the cellular microenvironment. These changes generate a precancerous niche (PCN), which is the end-product of fibrosis, remodeling secondary to persistent inflammation, and the deployment of a chronic stress escape strategy (CSES). Part of this Fig. was published with modifications from the original illustration from [35]. The lower drawn (Fig. 2b) is a cutout of the lamina fibroreticularis layer with additional fibroblasts that produce intracellular procollagen alpha-chains and release extracellular helical collagen trimers and mature procollagen. Ongoing disruption of homeostatic crosstalk results in the continuous increase in lysyl oxidase (LOX) activity. This result in increased oxidation of lysine residues in collagen leading to increased stiffness and decreased elasticity. Common abbreviations are in bold text, followed by the common trivial names and the International Union of Pure and Applied Chemistry (IUPAC) designations (as available): PCN, precancerous niche; CSES, chronic stress escape strategy; SphK, sphingosine kinase isoform; S1P, sphingosine-1-phosphate; IL-6, interleukin 6; IL-8, interleukin 8; TNFα, tumor necrosis factor alpha; IFNγ, interferon gamma; ALOX, lipoxygenase, arachidonate lipoxygenase; ALOX12, 12-lipoxygenase, 12-LOX, 12S-LOX, arachidonate 12-lipoxygenase 12S type; ALOX5, 5-lipoxygenase, 5-LOX, arachidonate 5-lipoxygenase; 12-HETE, 12-hydroxyeicosatetraenoic acid; LTA4 leukotriene A4, 4-[(2S,3S)-3-[(1E,3E,5Z,8Z)-tetradeca-1, 3,5, 8-tetraenyl]oxiran-2-yl]butanoic acid; LTB4, leukotriene B4, (5S,6Z,8E,10E,12R,14Z)-5, 12-dihydroxyicosa-6, 8,10, 14-tetraenoic acid; LTC4, leukotriene C4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-[[(4S)-4-amino-4-carboxybutanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-5-hydroxyicosa-7, 9,11, 14-tetraenoic acid; LTD4 leukotriene D4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-amino-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-5-hydroxyicosa-7, 9,11, 14-tetraenoic acid; LTE4, leukotriene E4, (5S,6R,7E,9E,11Z,14Z)-6-[(2R)-2-amino-2-carboxyethyl]sulfanyl-5-hydroxyicosa-7, 9,11, 14-tetraenoic acid; 5-oxo-ETE, (6E,8Z,11Z,14Z)-5-oxoicosa-6, 8,11, 14-tetraenoic acid; Cox, cyclooxygenase; Cox-1, cyclooxygenase 1; Cox-2, cyclooxygenase 2; Cox-3 isoform of Cox-2 (in brackets); PGG2, prostaglandin G2, (Z)-7-[(1S,4R,5R,6R)-5-[(E,3S)-3-hydroperoxyoct-1-enyl]-2, 3-dioxabicyclo [2.2.1]heptan-6-yl]hept-5-enoic acid; PGH2, prostaglandin H2, (Z)-7-[(1S,4R,5R,6R)-5-[(E,3S)-3-hydroxyoct-1-enyl]-2, 3-dioxabicyclo [2.2.1]heptan-6-yl]hept-5-enoic acid; PGF2α, prostaglandin F2 alpha, (Z)-7-[(1R,2R,3R,5S)-3, 5-dihydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]cyclopentyl]hept-5-enoic acid; PGD2, prostaglandin D2, (Z)-7-[(1R,2R,5S)-5-hydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxocyclopentyl]hept-5-enoic acid; PGE2, prostaglandin E2, (Z)-7-[(1R,2R,3R)-3-hydroxy-2-[(E,3S)-3-hydroxyoct-1-enyl]-5-oxocyclopentyl]hept-5-enoic acid; MDA, malondialdehyde, propanedial; TXA2, thromboxane A2, (Z)-7-[(1S,2S,3R,5S)-3-[(E,3S)-3-hydroxyoct-1-enyl]-4, 6-dioxabicyclo [3.1.1]heptan-2-yl]hept-5-enoic acid; CYP*, cytochrome P450 isoforms; 20-OH-PGE2, 20-hydroxy prostaglandin E2; 20-HETE, 20-hydroxyeicosatetraenoic acid, (5Z,8Z,11Z,14Z)-20-hydroxyicosa-5, 8,11, 14-tetraenoic acid; SOX, [sex-determining region Y (Sry) box-containing] transcription factor family; IL-β1, interleukin beta 1; IL-33, interleukin 33; ROS, reactive oxygen species; CXC CC, chemokine receptors; TNF, tumor necrosis factor alpha; αSMAD, alpha-smooth muscle actin; miR21, micro RNA-21; p300, protein 300 (p300-CBP coactivator family); SP1, specificity protein 1; AP1, activator protein 1; E2F4/5, cytoplasmic complex of Smad3, retinoblastoma-like protein 1 (P107, RBL1), E2F4/5 and d-prostanoid (DP1); p107, retinoblastoma-like protein 1, RBL1; LOX, lysyl oxidase; TGFβ, transforming growth factor beta; Pro-MMP-9, pro-matrix metalloproteinase 9; Pro-MMP-1, pro-matrix metalloproteinase 1; Pro-MMP-7, pro matrix metalloproteinase 7; SNAIL, zinc finger protein SNAI1; MMP-1, matrix metalloproteinase 1; MMP-7, matrix metalloproteinase 7; MMP-2, matrix metalloproteinase 2; E-Cadherin, CAM 120/80 or epithelial cadherin, cadherin-1, epithelial cadherin; PI3K, phosphatidylinositide 3-kinase; FOXO3a, forkhead box protein O3a; p120, catenin delta-1, protein 120; Rho, Ras homolog gene family, member A; Rac1, Ras-related C3 botulinum toxin substrate 1; cdc42, cell division control protein 42 homolog; BIM, Bcl-2 interacting mediator of cell death; PUMA, BH3-only protein; CXCR4, C-X-C motif of chemokine receptor 4.
Increased stromal rigidity induces mechanotransduction and is associated with poor survival in most cancers. LOXL2 and TGF-β1 receptor inhibition by trihydrophenolics attenuated lung and cancer fibrosis [38] while various signaling pathways and lysyl oxidase isoforms need to be considered. Of particular note, activation of the isoform, lysyl oxidase-like-2 (LOXL2) will result in decreased E-cadherin and increases fibronectin, vimentin, and metalloproteinases, and thus explains at least in part how the vicious cycle of ongoing chronic inflammation promotes the development of the PCN via the induction of lamellipodia and consequently in increased cell mobility and migration.
In a recent series of experiments, Wahlsten et al. [77] observed that a three-fold increase in collagen hydrogel stiffness-induced fibroblast maturation and proliferation. Fibroblasts at different stages of maturation can be detected simultaneously in the same organism including those with pronounced functional and structural variations with behaviors that are similar to embryonic cells [78–80]. Each cell is unique because all may be at different and distinct points in their maturation process; this results in general heterogeneity throughout [81–83]. As in all cells, fibroblast gene expression patterns are maturation-dependent [84]. Cells at different stages of maturation and development have distinct propensities for transformation and thus exhibit considerable heterogeneity [85].
For example, while fibroblasts can round up and stop migrating, they can eventually recover and move again [86]. Fetal fibroblasts and fibroblasts isolated from cancer patients actively express migration stimulating factor (MSF). This may explain why fibroblasts from cancer patients exhibit fetal-like migratory behavior and increased cell density while fibroblasts isolated from patients with more benign diseases do not [87]. Furthermore, epithelial cells can extend basal lamellipodia that facilitate active crawling [88], as discussed in depth in an earlier study [89].
Fibroblast-specific protein-1 (FSP1, S100A4) was initially identified exclusively in fibroblasts and was not detected in epithelial cells [90]. De novo expression of FSP1 is one of the critical events associated with early EMT; others include up-regulation of vimentin expression and reductions in cytokeratin [91]. However, FSP1 expression in this setting does not rule out the existence of additional cellular sources; for example, FSP1 has been found in CD4+ and CD8+ T-lymphocytes (but not B-lymphocytes), subendothelial smooth muscle cells as well as those in the tunica adventitia and tunica media, epithelial cells in the kidney and the bladder, the stroma of the prostate, adipocytes, and skeletal muscles [92]. Among its activities and functions, FSP1 promotes EMT, fibrosis, pulmonary vascular disease, metastatic tumor development, and increased tumor cell motility and invasiveness.
During renal fibrosis, fibroblasts are derived primarily from the renal epithelium via a process of cell transition and in smaller amounts from cell migration from the bone marrow [93]. Stromal cells migrating from bone marrow that can develop a fibroblast phenotype in tissues are an important source of fibroblasts available for recruitment [94]. One of the consequences of ongoing collagen production and LOX-mediated cross-linking is a substantial increase in stromal rigidity and associated reduced elasticity (FIGURE 2).
Stiffness, as indicated by the parameter κ, is defined as the resistance to deformation (δ) when subjected to an applied force (Ϝ). This relationship can be described by the one-degree model written as κ = Ϝ / δ. This is the formula for a basic elastic model, for example, one that can be used to represent a simple coil spring. However, this concept becomes somewhat more complicated if more than one-degree of force is applied to an object and if one needs to model responses in a three-dimensional space (i.e., the extracellular matrix). In this case, the stiffness of the matrix will depend on various dimensional forces, including the elastic modulus, which is a property of the constituent material. If the properties of the matrix and the material present different states of aggregation and tensions at the same time (i.e., the physical state), the complexity increases further. Johannes Diderik van der Waals (1837–1923) described the dispersive forces (i.e., van der Waals forces) that control the interactions of the low-energy surfaces found between liquids. As we can see, an understanding of this concept is critical for an appreciation not only of physics but also of human biology.
The stiffness concept was introduced to explain how durotactic velocity increases with the stiffness gradient as well as to highlight the differences between active traction at the front and in the rear ends of the cell [95]. The term durotaxis, which is defined as cell migration that depends on mechanical factors, was first introduced to explain the behavior of neurites in cell culture [96] and has since been used to describe cell migration in general [97]. Maximal cell motility occurs at intermediate stiffness and durotactic velocity increases with cluster size, the stiffness gradient, and actomyosin activity [95]. This concept helps us to understand cancer cell migration and its responses to these parameters. For example, cancer cluster cell velocity is minimal at low stiffness (0.2 kPa), peaks at intermediate stiffness (24 kPa), and decreases at high stiffness (200 kPa). However, each organ and tissue exhibits a characteristic physiological stiffness (within the limits of homeostasis). ECM stiffness and the speeds of cell migration depend on contractility mediated by non-muscle myosin II (NMMII) [98]. In Pallares et al. [95], the authors theorized that ”…local maximum velocity may theoretically exist on significantly stiffer ECMs (i.e., > 150–200 kPa).” Since that time, others have shown that fibrosis induces increases in ECM stiffness (120 kPa) and thus promotes EMT [99].
It is important to recognize that fibrosis is a wound-healing repair mechanism and thus an important part of physiological homeostasis; here, LOX inhibits fibroblast transformation as part of a negative loop. However, ongoing disruption of homeostasis secondary to an unresolved harmful stimulus and chronic inflammation will promote a pathological increase in stiffness. As a result, initial efforts to adapt and achieve allostasis involve fibroblast recruitment and thus increased production of intracellular alpha chains. This is accompanied by procollagen release to the extracellular space and the creation of helical trimers and mature tropocollagen. Increases in LOX activity will result in increased oxidation of lysine residues which will result in pathological collagen cross-linking and consequently increased stiffness and reduced elasticity. Increases in matrix stiffness will facilitate cancer cell transition.
Cell transition
Cell transition is a universal process that occurs in embryogenesis, tissue maturation and differentiation, and cell turnover, and which can promote imbalanced homeostasis. In humans, 10 to 50 million cells turnover each second [100, 101]. The average lifespans of various cells and cell types differ significantly [102–105]. Notable differences are observed in epithelial cells, including those of the gastric cardia (average lifespan of 9.1 days), gastric pylorus (1.8 days), small bowel (1.4 days), colon (10 days), rectum (1.4 days), anus (4.3 days), and peritoneal wall (19.4 days). These values can be compared to those reported for the glandular epithelia, including cells in the liver (222 days), kidney (286 days), and thyroid (287 days). Furthermore, both EMT and mesenchymal-epithelial transition (MET) can be reversed in the presence of TGF-β [106].
Fibrosis induces signaling and crosstalk leading to mechanotransduction and consequently EMT [106–113]. ECM stiffness, including cell-matrix adhesions and cytoskeletal polarization, also induces EMT [99]. Decreased stiffness has been associated with inhibited EMT that is not dependent on apoptosis [114]. Increased levels of vimentin and decreased levels of E-cadherin and β-catenin are observed in the presence of increased ECM stiffness (e.g., 120 versus 20 kPa) [99], leading to elevations in chronic tissue stress. One of the consequences of this scenario is the chronic stress escape scenario (CSES) followed by normal cell-to-cancer cell transition (NCCCT).
Cell transition has been reported in all histological types of cancer, including epithelial cancers [115, 116], sarcomas [117, 118], leukemias [119–123], lymphomas [124], myelomas [125, 126], and central nervous system cancers [127, 128].
Cell transition toward pluripotency occurs in dermal fibroblasts undergoing maturation. After seven days, 20% of the cells express the pluripotent stem cell marker TRA-1-60, although only 1% generate colonies of induced pluripotent stem cells after replating; most of the cells return to the original TRA-1-60-negative state [129]. As more fibroblasts mature, the percentage that reverts to the original state decreases under normal conditions.
Molecular mechanisms underlying EMT and MET
The process known as EMT was first described in 1982 [130, 131]. Many recent studies have focused on the process via which cancer cells undergo EMT and why circulating epithelial cells exhibit mesenchymal characteristics [132]. Epithelial cell transition results in cells with disrupted apicobasal polarity and disassembled cell-to-cell junctions that lead to impaired epithelial layer integrity and loss of its barrier function [133]. These findings also explain why the resulting cell phenotype includes a higher capacity for migration with cells moving away from their normal positions. Among these changes, cells that have undergone EMT can lose their solid-type morphology and acquire a fluid-like migratory phenotype; this is known as “unjammed transition” (UJT) [134]. The term “jamming” represents a collective invasion strategy used by cancer cells [135]. The results of several investigations have clarified this mechanism [134], and also yielded the term “unjamming cell transition” to reflect the responses of epithelial cells as they change from a silent to a migratory condition and undergo UJT [136 reviewed in 133].
Small ubiquitin-related modifiers (SUMOs) have a profound influence on cell transition. SUMOs are a family of proteins that are structurally similar to ubiquitin but exhibit distinct amino acid sequences and functions [137]. SUMO-1, SUMO2, and SUMO-3, are three distinct structural forms that bind to form covalent or non-covalent isopeptide bonds with ɛ-amino groups of acceptor Lys residues, a process known as “SUMOylation” [138–143]. At current writing, five isoforms of SUMO have been identified in the human genome [144].
SUMO biosynthesis and activation require multiple specific steps. SUMO protein undergoes proteolysis and becomes attached to a heterodimeric E1 complex (SAE1/UBA2). The energy for this reaction is provided by the ATP-dependent activating enzyme (E1). The SUMO peptide is subsequently transferred to the central E2 enzyme (UBC9); an E3 ligase attaches it to a target protein. Afterward, deconjugation occurs via the actions of SUMO proteases [144–147].
SUMO has been recognized as a post-translational modifier in hundreds of eukaryotic cells with metabolism-influenced signaling and crosstalk pathways. While SUMO-1 decreases autophagy [148, 149], it also significantly increases hypoxic cell stress [150], and induces the expression of Ras-related C3 botulinum toxin substrate 1 (RAC1) [149]. Collectively, this leads to microtubule polymerization and integrin clustering that will progress to lung cancer [151, 152], glioblastoma [153], endometrial cancer [154], actinic cheilitis and lip carcinoma [155], liver cancer [156, 157], gastric cancer [158] or pancreatic carcinoma [159]. SUMOylation is used by viruses to modify their own proteins [160, 161], most notably by HPV [162], cytomegalovirus (CMV) [163) or Epstein–Barr virus (EBV) [164, 165] to support their own replication.
The SUMO-specific protease 2 (SENP2) catalyzes deSUMOylation, thereby suppressing TGF-β-induced EMT [166]. Likewise, fibrosis is prevented/attenuated from inhibiting SUMOylation secondary to a decrease in TGF- signaling [167]. However, SUMOylation is only one of the regulatory processes that promotes or inhibits various intracellular signaling pathways [168–170].
EMT and MET are mechanisms that explain cell migration, invasiveness, and metastasis and thus make pivotal contributions to the process of carcinogenesis. Cancer-associated fibroblasts (CAFs) also play a significant role in this process and are already present in precancerous lesions.
Cancer-associated fibroblasts (CAFs)
Precancerous tissues exhibit increased stromal rigidity and reduced elasticity. Cancer-associated fibroblasts (CAFs) have been detected in these lesions, including those leading to colorectal adenoma [171–174], ulcerative colitis [175], Barrett’s esophagus [176], atrophic gastritis [177], leukoplakia [178, 179], and pituitary adenoma [180]. CAFs have also been identified in precancerous lesions of the blood, for example, monoclonal gammopathy of unknown significance [181-183] and the breast. Tenascin, which is a marker for CAFs [184], has also been detected in precancerous breast lesions as well as in ductal or lobular carcinoma in situ [185–193].
While CAFs have been detected in epithelial cancers, they also promote carcinogenesis in sarcomas [194–196], leukemias [197–201], lymphomas [202-205], myelomas [183, 206] and central nervous system cancers [207–210]. CAFs exhibit considerable heterogeneity [211, 212]. Conversion of fibroblasts to CAFs has also been demonstrated [213].
When compared to their noncancerous counterparts, CAFs from human breast tumors exhibit greater up-regulation of NADPH oxidase 4 (Nox4) than epithelial cells. In addition, CAFs in breast tumors are associated with increased autophagy and survival via increased levels of nuclear factor erythroid 2-related factor 2 (Nrf2, also known as NFE2L2, Heme Binding Protein 1, HEBP1, nuclear factor, erythroid 2 like 2, IMDDHH, Nrf-2, NFE2 like bZIP transcription factor 2). Nox4 deactivation reverses these changes [214], which is consistent with findings resulting from a decrease in TGFβ-mediated fibroblast activation [215]. Nrf2 was first isolated in 1994 [216] and has since been recognized as a dormancy marker in human breast cancer cells [217].
Administration of the aryl hydrocarbon receptor (AHR) agonist, 2, 3,7, 8-tetrachlorodibenzo-p-dioxin (TCDD), results in increased collagen-type I (Col1A1), α-smooth muscle actin, and pro-inflammatory cytokine synthesis and decreased E-cadherin and claudin 1. These responses are associated with fibrosis upregulation and cell transition via AHR-mediated epidermal growth factor receptor/extracellular signal-related kinase (EGFR/ERK) signaling pathways [41] and induce AHR-mediated expression of Nrf2 [218]. Lung CAFs produce the tryptophan metabolite known as kynurenine (Kyn) which also increases AHR [219], and vimentin expression and AHR translocation [41]. AHR-mediated signaling induces EMT [220]; by contrast, cytoplasmic translocation of the AHR suppresses EMT [221]. Selective AHR blockade with 3, 3'-diindolylmethane (DIM) inhibits the Ras homolog gene, member A/rho-associated, coil-containing protein kinase 1 (RhoA/ROCK1), which modulates the cyclooxygenase-2/prostaglandin E2 (COX2/PGE2) pathway connected to nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); this will result in EMT reversal [222].
The protein p120-catenin (p120[ctn]) can typically be detected at the membrane bound to E-cadherin. This is accompanied by Rho GTPase-Cdc42 signaling, which is important for membrane stability [223, 224]. This is accompanied by the loss of E-cadherin; loss or dysfunctional E-cadherin/catenin results in the redistribution of cytoplasmic p120 [225, 226]. An increase of N-cadherin expression together with a relocalization of N-cadherin, α-catenin, p-120, and β-catenin, will lead to fibroblast transformation into a cell with epithelial-like morphology [227]. The contributions of E-cadherin to functional homeostasis are important because these cells control cellular motility as well as p120ctn overexpression in fibroblasts. Results of transfection studies revealed that increased filopodial/lamellipodial activity in these cells was mediated by small guanosine triphosphatases (GTPases) of the Rho family [228, 229]. Here, the increase of N-cadherin, the loss of E-cadherin, and the expression of p120 promote an EMT phenotype in the surrounding fibroblasts [230]. Furthermore, p120 expression is a co-regulator of the fibroblast cell cycle and exhibits an increased G1 to S phase [231]. Fibroblasts exposed to a pathogen, for example, Abelson murine leukemia virus (MuLV), generate TGF release and an increase in p120 protein [232].
Kaiso is a bimodal transcriptional repressor that contains both a zinc‐finger and a BTB/POZ domain [233] that has been associated with protective effects in triple-negative breast cancer [234]. By contrast, nuclear Kaiso has been associated with clinically aggressive breast cancer [235]; nuclear localization may promote both an aggressive phenotype and EMT [236, 237]. Kaiso phosphorylation leads to a functional switch toward oncogenesis by promoting cancer cell growth in vivo [238]. This results in cytoplasmic accumulation and binding to p120 and 14-3-3 family proteins and will ultimately lead to the translocation of Kaiso from the nucleus into the cytoplasm and a negative feedback loop associated with the Kaiso target gene, cadherin 1 (CDH1) in normal tissues. This largely explains why cytoplasmic Kaiso is associated with poor prognosis in lung cancer [239]. Kaiso also promotes EMT via its capacity to regulate miR-200c [240]; by contrast, miRNA-4262 upregulation inhibits EMT [241].
Silencing of p120 decreases fibrosis [242] and activates p120/Kaiso in the absence of Wnt/Smad/ZEB signaling, thereby preventing EMT [243]. Fibroblast and surface protein markers are expressed on both benign and malignant fibroblasts and epithelial cells [244]. Marker expression is not fibroblast or CAF specific, but rather is associated with the acquisition of the mesenchymal phenotype by epithelial cells during EMT.
Consequence
Immunosuppression is a common feature of disrupted homeostasis observed in cancer; CAFs become dominant in this setting. These findings are supported by gene co-expression data [245]. Histopathologically, airway epithelial cells that have undergone EMT have also been detected in immunodeficient patients who have undergone organ transplantation [246].
Persistent disruption of homeostatic crosstalk generates a precancerous niche (PCN) with ongoing fibrosis and remodeling by LOX; the CSES deploys EMT to avert and resolve this situation. In this setting, the microenvironment remains under increasing chronic stress with continuous increases in stiffness due to fibroblast collagen production and LOX activity [28, 29, 33–43]. This amplifies fibroblast activation which results in increased collagen production and cross-linking. This also results in the induction of IL-6 and activator protein 1 (AP-1) which leads to inflammation and stroma production. Of note, LOX activates ~134 genes expressed by fibroblasts [247]. Further increases in fibronectin, vimentin, and metalloproteinases together with decreases in E-cadherin explain why epithelial cell separation can occur more readily under these conditions. EMT, as well as the recruitment of fibrocytes and fibroblasts from the bone marrow collectively explains the extent of fibroblast heterogeneity observed under these conditions. Here, epithelial cells undergo EMT to fibroblasts, which will contain epithelial markers (FIGURE 3). As chronic inflammation persists and fibrosis increases, many signaling and crosstalk pathways are activated. This creates a favorable microenvironment in which affected cells become more susceptible to neoplastic transformation [28, 29, 33–43].
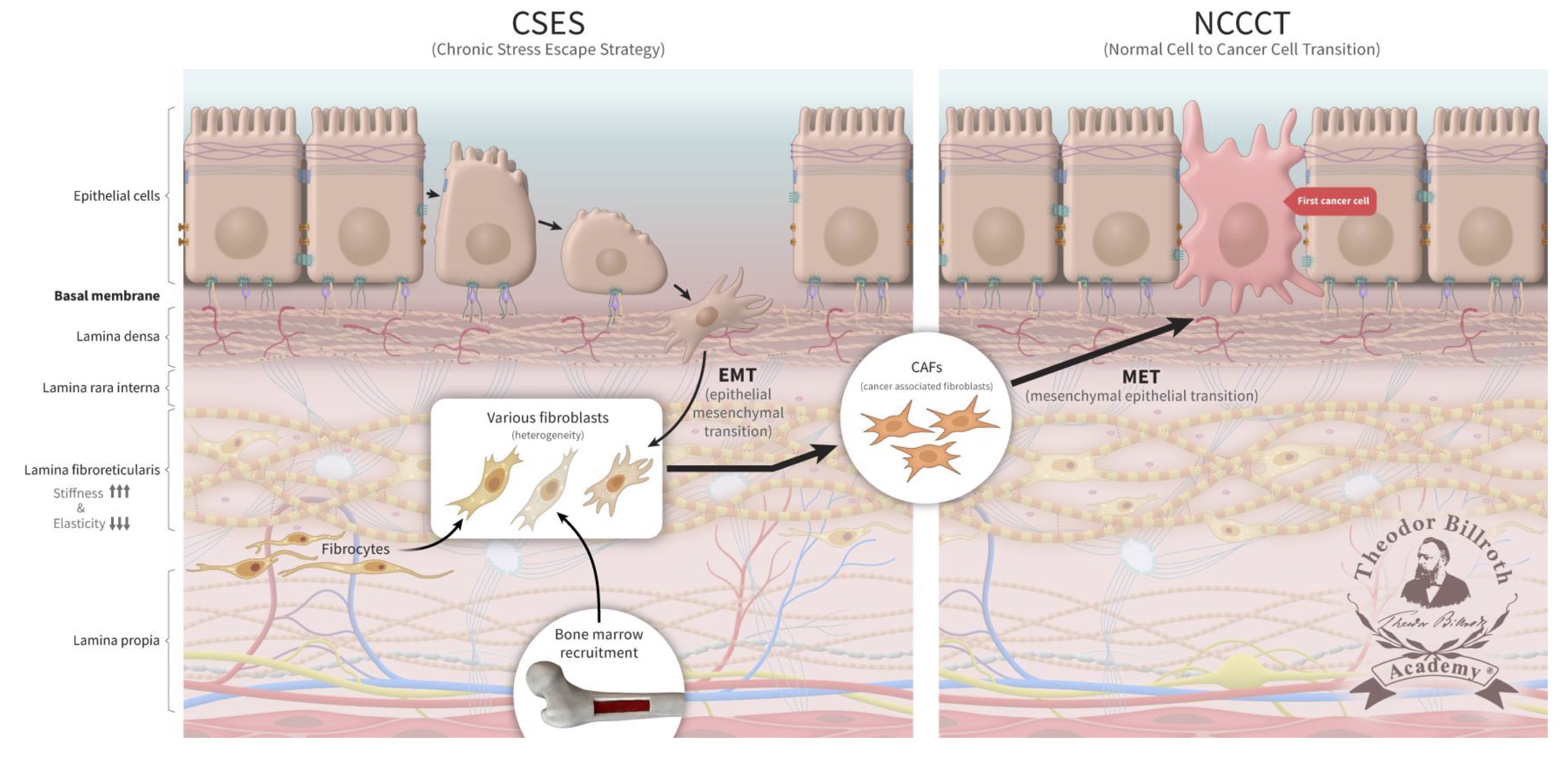
Fig. 3: The chronic stress escape strategy (CSES) deployed in a precancerous niche (PCN) followed by normal cell-to-cancer cell transition (NCCCT). Unresolved, chronic tissue stress in a precancerous niche (PCN) is characterized to lead to an escape strategy, chronic stress escape strategy (CSES), that involves the recruitment of fibroblasts and fibrocytes from the bone marrow as well as cells undergoing an epithelial-mesenchymal transition (EMT). This yields a heterogeneous pool of cells that express both epithelial and mesenchymal markers that will ultimately differentiate into cancer-associated fibroblasts (CAFs) in the PCN. The persistent disruption of homeostatic crosstalk increases lysyl oxidase activity and lysine oxidation which will lead to increased collagen stiffness and decreased elasticity. Finally, CAFs undergo a mesenchymal-epithelial transition (MET) and express epithelial markers that facilitate their integration into the target tissue. The continuous increase in CAFs represents the final step that leads to the complete and unresolvable disruption of physiologic homeostasis. CAFs then undergo MET; these cells, which continue to express epithelial markers, and become the first cancer cells. These former fibroblasts are then integrated into the epithelium (FIG. 3).
Results of experiments performed in the blind mole rat, Spalax, which is a long-lived mammal that is resistant to chemical carcinogenesis and cancer reveal that their fibroblasts can suppress colony formation in human breast cancer cell lines [248–250]. Unlike human or mouse fibroblasts, Spalax fibroblasts can promote continuous downregulation of inflammatory signals and thus can limit aging-associated chronic inflammation [251].
Considering there is very high heterogeneity between different cancer types, another question is, if the heterogeneity between different cancer types is observed after the initial cell is transformed or before. Much (or all) of the observed heterogeneity observed in different cancer types are related to factors that include a) stage of cancer at the time of observation; b) morphology associated with organ containing the tumor, e.g., lung, liver, breast, colon, etc., and c) spread of the tumor within the affected organ. Based on our understanding and of that of the literature, and in the context of the cancer paradigm "Epistemology of the Origin of Cancer" [28–43], the earliest changes that occur in the transformation of a normal cell to a cancer cell must be common to most solid cancers and this "first cancer cell" is a fibroblast, not an epithelial cell though later in the process of carcinogenesis, cancer cells "appear" as an epithelial type.
Currently, cancer tissues collected during a biopsy or surgery are fixed and reviewed by a pathologist who performs a microscopic evaluation to determine the cell type and morphology. These methods continue to have a powerful influence on the choices of cancer-specific therapeutics. Of note, most cancerous tissues obtained in this fashion will exhibit an epithelial morphology. This may have led to the assumption that most cancer cells must originate from once healthy epithelial cells. As demonstrated, the cells that appear to be epithelial and that express epithelial markers may have been fibroblastic in origin. In this manuscript, we provide evidence suggesting that fibroblasts serve as the initial precursors to these cancer cells.
Conclusion
Retrospectively, we may wonder why physiological observations that provided us with the information needed to address this unsolved puzzle were unseen though right in front of our eyes. However, we may also recognize that the physiological complexity was such that these strands could not be woven together. As stated by Friedrich Schiller in 1796 (English translation) “What’s the hardest of all? What you think is the easiest, to see with your eyes what’s in front of your eyes” [252]. It was not by accident that Professor Frithjof Hammersen (1889–1984) included this quote in his famous atlas of histology [253].
In regard to our today’s fibroblasts knowledge, we stand on the shoulders of giants, among them, Carl Bogislaus Reichert (1811–1883) [254], Hermann Lebert (1813–1878) [255], Rudolf Virchow (1821–1902) [256], Wilson Fox (1831–1887) [257], Ernst Ziegler (1849–1905) [258], William Russell (1852–1940) [259], Santiago Ramón y Cajal (1852–1934) [260], Leo Loeb (1869–1959) [261], and Alexander Alexandrowitsch Maximow (1874–1928) [262].
While the importance of fibroblasts and their contributions to carcinogenesis was suggested long ago (Supplement Part 3, Fibroblasts: historical consideration), epithelial cells are still believed to be the primary cell in this process, even in carcinoma in situ. While this is understandable, given that these are the first cells detected based on microscopic techniques typically used for diagnosis, it is important to have a sense of what changes have occurred previously. As discussed in this manuscript, accumulated evidence points to the fibroblast as the first cell that truly gives rise to cancer.
The findings illustrated in FIGURE 3 outline a complex but persuasive argument for the identification of the fibroblast as the first cancer cell. These findings also explain the considerable heterogeneity detected in the precancerous niche (PCN), which contains various types of stromal cells, cancer cells, and resected cancer masses. During cancer development, progression, differentiation, cell transition, and cell division, ongoing EMT followed by MET is also observed. This explanation provides insight into the many factors that may undermine therapies, including cell resistance (based on selection pressure, mutational burden, and expression of multidrug exporter pumps) and the extent of vascularization. Likewise, the low proliferative index of specific cancer cells will limit their susceptibility to cytotoxic chemotherapy. Multimodal therapy approaches alone are unlikely to result in a substantial difference when considering the most important issues for cancer patients, namely, increased survival measured in years rather than months or weeks.
Effective prophylactic measures and potential cures for cancer as well as prophylaxis will only emerge from an understanding of the correct etiology of this disease. Thus, further exploration is needed to define and characterize the sequence of events leading to the development of most cancers [28, 29, 33–43] and, as provided here, to formulate a plausible explanation of the origins of cancer together with a clear identification of the precursor cells that transition to cancer via the sequence of events defined as described above. Thus, we are now in a position to develop new anticancer strategies that target the precancerous niche (PCN); these and other rational therapies may lead us toward the original vision of “Imagine a world without cancer” [32].
Disclosure Statement
The authors report no Disclosure Statement. The authors alone are responsible for the content and writing of this manuscript. This manuscript contains original material that has been previously published and is appropriately cited. The opinions or assertions contained herein are those of the authors alone and are not to be construed as official or reflecting the views of the publisher or their employers. Both authors contributed to the content and approved the final version of the manuscript.
Abbreviations
AHR, aryl hydrocarbon receptor; AP-1, activator protein 1; BPAG1, bullous pemphigoid antigen 1; CAFs, cancer-associated fibroblasts; CD44, cell surface receptor glycoprotein; CD151, cluster of differentiation 151; CDH1, cadherin 1; CMV, cytomegalovirus; CNC, cap`n collar family of basic leucine zipper transcription factors; Col1A1, collagen-type I; Col3, collagen-type III; Col4, collagen-Type IV; Col7, collagen type VII; Col17, collagen type XVII; COX2/PGE2, cyclooxygenase-2/prostaglandin E2; CSES, chronic stress escape strategy; DIM, 3, 3'-diindolylmethane; E1, ATP-dependent activating enzyme; EBV, Epstein–Barr virus; ECM, extracellular matrix; EGFR/ERK, epidermal growth factor receptor/extracellular signal-related kinase; EMT, epithelial-mesenchymal cell transition; EndMT, endothelial–mesenchymal transition; FSP1, fibroblast-specific protein-1, S100A4; GTPase, guanosine triphosphatase; HSPG2, heparan sulfate proteoglycan 2, perlecan; HPV, human papilloma virus; iPS cells, pluripotent stem cells; Kyn, kynurenine; LOX, lysyl oxidase; LOXL2, lysyl oxidase-like-2; MET, mesenchymal-epithelial cell transition; MSF, migration stimulating factor; MuLV, murine leukemia virus; NCCCT, normal cell-to-cancer cell-transition; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nidogen-1, entactin; NMMII, non-muscle myosin II; Nox4, NADPH oxidase 4; Nrf2, nuclear factor erythroid 2-related factor 2; p120(ctn), p120-catenin; PCN, precancerous niche; RAC1, Ras-related C3 botulinum toxin substrate 1; RhoA/ROCK1, Ras homolog gene, family, member A/rho-associated, coiled-coil-containing protein kinase 1; RSV, Rous sarcoma virus; SENP2, SUMO-specific protease 2; SUMO, small ubiquitin-related modifiers; TβR , TGF-β1 receptor; TCDD, 2, 3,7, 8-tetrachlorodibenzo-p-dioxin; TGF-β1, tumor growth factor beta; TRA-1-60, pluripotent stem cell marker; UBC9, central E2 enzyme; UJT, unjammed transition.
Acknowledgements
We acknowledge the independent critical pro bono peer review provided to us by Professor Dr. Marjan Slak Rupnik, Physiologist & Pathophysiologist, Vienna, Austria (ORCID: https://orcid.org/0000-0002-3744-4882); Professor Dr. Detlef Bartsch, Head Genetic Engineering, German Federal Office for Consumer Protection and Food Safety in Berlin, Germany (ORCID: https://orcid.org/0000-0003-2961-915X); Dr. Björn Petersen, Expert for Xenotransplantation, Friedrich-Loeffler-Institut, Institute of Farm Animal Genetics in Neustadt, Germany (ORCID: https://orcid.org/0000-0002-1532-4863); Dr. Romain-Daniel Gosselin, Physiopathologist, Lausanne University Hospital, Lausanne, Switzerland (ORCID: https://orcid.org/0000-0003-1716-6210); Dr. Gudrun Schueler, Hematologist & Oncologist, Cottbus, Germany; Professor Dr. Jose Florencio F Lapeña Jr., Otolaryngologist, World Association of Medical Editors, Manilla, the Philippines (ORCID: https://orcid.org/0000-0002-5794-1878). Each of these individuals has approved this acknowledgment. Funding: Partial funding that enabled us to purchase non-open access publications necessary for this research was granted by the Theodor-Billroth-Academy®, Munich, Germany, and Sacramento, CA, USA. Author contributions: BB wrote the first draft of the manuscript and IJ contributed to specific sections. Both authors edited and modified the manuscript. This manuscript was created from our intellectual input only. No artificial intelligence software, e.g., ChatGPT or others was used in the preparation of this manuscript.
References
| 1 | Rous P, Murphy JB: Variations in a chicken sarcoma caused by a filterable agent. J Exp Med 1913;17:219-231.
https://doi.org/10.1084/jem.17.2.219 |
| 2 | Rous P, Murphy JB: On the causation by filterable agents of three distinct chicken tumors. J Exp Med 1914;19:52-68.
https://doi.org/10.1084/jem.19.1.52 |
| 3 | Levin I: Tumor onoculation into organs and the analogy between human cancer and the tumors of white mice and white rats, J Exp Med 1912;16:155-164.
https://doi.org/10.1084/jem.16.2.155 |
| 4 | Smith WE, Rous P: The neoplastic potentialities of mouse embryo tissues: II. Contributory experiments; results with the skin of C3H and Webster-Swiss embryos; general considerations. J Exp Med 1945;81:621-646 .
https://doi.org/10.1084/jem.81.6.621 |
| 5 | Teperson JA, Altman LS, Kogut B: Multiple heterogeneous carcinoma of the stomach. J Int Coll Surg 1952;17:374-378.
|
| 6 | Potter VR: Biochemical uniformity and heterogeneity in cancer tissue (further discussion), Cancer Res 1956;16:658-667.
|
| 7 | Kit S: Compositional heterogeneity of normal and malignant tissue deoxyribonucleic acids (DNA), Biochem Biophys Res Commun 1960;3:361-367.
https://doi.org/10.1016/0006-291X(60)90045-0 |
| 8 | Fahey JL: Physiocochemical characterization of mouse myeloma proteins: demonstration of heterogeneity for each myeloma globulin. J Exp Med 1961;114:399-413.
https://doi.org/10.1084/jem.114.3.399 |
| 9 | Fidler IJ, Kripke ML: Metastasis results from preexisting variant cells within a malignant tumor. Science 1977;197:893-895.
https://doi.org/10.1126/science.887927 |
| 10 | Fidler IJ: Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res 1978;38:2651-2660.
|
| 11 | Fidler IJ (2012) Biological heterogeneity of cancer: implication to therapy. Hum Vaccin Immunother 8:1141-1142.
https://doi.org/10.4161/hv.19643 |
| 12 | Fidler IJ: Commentary on "Tumor Heterogeneity and the Biology of Cancer Invasion and Metastasis". Cancer Res 2016;76:3441-3442.
https://doi.org/10.1158/0008-5472.CAN-16-1330 |
| 13 | Murphy JB, Sturm E: Effect of stimulation of the lymphocytes on the rae of growth of spontaneous tumors in mice. J Exp Med 1919;29:31-34.
https://doi.org/10.1084/jem.29.1.31 |
| 14 | Nakahara W; Effect of fatty acids in the resistance of mice to transplanted cancer. J Exp Med 1924;40:363-373.
https://doi.org/10.1084/jem.40.3.363 |
| 15 | Bertino JR: Third Myron Karon Memorial Lecture. Resistance of human tumors to cancer chemotherapeutic agents: an important research problem. Med Pediatr Oncol 1978;5:105-114.
https://doi.org/10.1002/mpo.2950050117 |
| 16 | Sun D, Feng F, Teng F, Xie T, Wang J, Xing P, Qian H, Li J: Multiomics analysis revealed the mechanisms related to the enhancement of proliferation, metastasis and EGFR-TKI resistance in EGFR-mutant LUAD with ARID1A deficiency. Cell Commun Signal 2023;21:48.
https://doi.org/10.1186/s12964-023-01065-9 |
| 17 | Hill BT, Douglas ID, Grover PL: Increased antitumour activity of chlorambucil following pretreatment with inducers of drug-metabolizing enzymes. Biochem Pharmacol 1973;22:1083-1089.
https://doi.org/10.1016/0006-2952(73)90173-1 |
| 18 | Flentje D, Schlag P: Is chemosensitivity testing for peri-operative treatment planning in gastro-intestinal cancer by the human tumour colony assay worthwhile? Eur J Surg Oncol 1985;11:227-233.
https://doi.org/10.1016/0305-7372(84)90052-5 |
| 19 | Lim SM, Mohamad Hanif EA, Chin SF: Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci 2021;11:56.
https://doi.org/10.1186/s13578-021-00570-z |
| 20 | Brennick CA, George MM, Corwin WL, Srivastava PK, Ebrahimi-Nik H: Neoepitopes as cancer immunotherapy targets: key challenges and opportunities. Immunotherapy 2017;9:361-371.
https://doi.org/10.2217/imt-2016-0146 |
| 21 | Jenks S: After the early hype, interferons spark interest. J Natl Cancer Inst 1993;85:773-775
https://doi.org/10.1093/jnci/85.10.773 |
| 22 | Jenks S: Panel says gene therapy "hype" should be toned down. J Natl Cancer Inst 1996;88:9-10.
https://doi.org/10.1093/jnci/88.1.9 |
| 23 | Chabanovska O, Galow AM, David R, Lemcke H: mRNA - A game changer in regenerative medicine, cell-based therapy and reprogramming strategies. Adv Drug Deliv Rev 2021;179:114002.
https://doi.org/10.1016/j.addr.2021.114002 |
| 24 | Xue VW, Wong SCC, Song G, Cho WCS: Promising RNA-based cancer gene therapy using extracellular vesicles for drug delivery. Expert Opin Biol Ther 2020;20:767-777.
https://doi.org/10.1080/14712598.2020.1738377 |
| 25 | Mosenhauer M, Newall PWS, Walasek L: The stock market as a casino: Associations between stock market trading frequency and problem gambling. J Behav Addict 2021;10:683-689.
https://doi.org/10.1556/2006.2021.00058 |
| 26 | Moore AE: Effect of inoculation of the viruses of influenza A and herpes simplex on the growth of transplantable tumors in mice. Cancer 1949;2:516-524.
https://doi.org/10.1002/1097-0142(194905)2:3<516::AID-CNCR2820020316>3.0.CO;2-P |
| 27 | Nahum LH: Viruses and cancer: herpes virus vaccine protests against Marek's disease of chickens. Conn Med 1970;34:398-399.
|
| 28 | Brücher BLDM, Jamall IS: Epistemology of the Origin of Cancer: A New Paradigm. BMC Cancer 2014;14:1-15.
https://doi.org/10.1186/1471-2407-14-331 |
| 29 | Brücher BLDM, Jamall IS: Cell-Cell communication in tumor microenvironment, carcinogenesis and anticancer treatment. Cell Physiol Biochem 2014;34:213-243.
https://doi.org/10.1159/000362978 |
| 30 | Brücher BLDM, Jamall JS: Somatic Mutation Theory - Why it's Wrong for Most Cancers. Cell Physiol Biochem 2016;38:1663-1680.
https://doi.org/10.1159/000443106 |
| 31 | Brücher BLDM, Yan L, Schnabel P, Daumer M, Wallace TJ, Kube R, Zilberstein B, Steele S, Jamall IS: Genomics, microRNA, epigenetics, and proteomics for future diagnosis, treatment and monitoring response in upper GI cancers. Clin Trans Med 2016;5:1-16.
https://doi.org/10.1186/s40169-016-0093-6 |
| 32 | Brücher BLDM, Lyman G, van Hillegersberg R, Pollock RE, Lordick F, Yang HK, Ushijima T, Yeoh KG, Skricka T, Polkowski W, Wallner G, Verwaal V, Garofalo A, D'Ugo D, Roviello F, Steinau HU, Wallace TJ, Daumer M, Maihle N, Reid III TJ, Ducreux M, Kitagawa Y, Knuth A, Zilberstein B, Steele SR, Jamall IS: Imagine a World Without Cancer. BMC Cancer 2014;14:1-8.
https://doi.org/10.1186/1471-2407-14-186 |
| 33 | Brücher BLDM, Jamall IS: Prelude and Premise to the Special Issue: Disruption of homeostasis-induced signaling and crosstalk in the carcinogenesis paradigm "Epistemology of the origin of cancer". 4open 2019;2:1-8.
https://doi.org/10.1051/fopen/2019005 |
| 34 | Brücher BLDM, Jamall IS: Undervalued ubiquitous proteins. 4open 2019;2:1-13.
https://doi.org/10.1051/fopen/2019010 |
| 35 | Brücher BLDM, Jamall IS: Chronic inflammation evoked by pathogenic stimulus during carcinogenesis. 4open 2019;2:1-22.
https://doi.org/10.1051/fopen/2018006 |
| 36 | Brücher BLDM, Jamall IS: Eicosanoids in carcinogenesis. 4open 2019;2:1-34.
https://doi.org/10.1051/fopen/2018008 |
| 37 | Brücher BLDM, Jamall IS: Microbiome and morbid obesity increase pathogenic stimulus diversity. 4open 2019;2:1-16.
https://doi.org/10.1051/fopen/2018007 |
| 38 | Brücher BLDM, Jamall IS: Precancerous niche (PCN), a product of fibrosis with remodeling by incessant chronic inflammation. 4open 2019;2:1-21.
https://doi.org/10.1051/fopen/2018009 |
| 39 | Brücher BLDM, Jamall IS: Metformin alters signaling homeostasis. 4open 2019;2:1-17.
https://doi.org/10.1051/fopen/2019006 |
| 40 | Brücher BLDM, Lang F, Jamall JS: NF-κB signaling and crosstalk in carcinogenesis. 4open 2019;2:1-35.
https://doi.org/10.1051/fopen/2019010 |
| 41 | Brücher BLDM, Jamall IS: Transition from normal to cancerous cell by precancerous niche (PCN) induced chronic cell-matrix stress, 4open 2019;2:1-31.
https://doi.org/10.1051/fopen/2018996 |
| 42 | Brücher BLDM, Jamall IS: Synopsis - Special Issue: Disruption of homeostasis-induced signaling and crosstalk in the carcinogenesis paradigm "Epistemology of the origin of cancer". 4open 2019;2:1-30.
https://doi.org/10.1051/fopen/2019023 |
| 43 | Brücher BLDM, Daumer M, Jamall IS: Physics Essentials Enable Deeper Understanding in Signaling and Crosstalk of the Carcinogenesis Paradigm "Epistemology of the Origin of Cancer". Cell Physiol Biochem 2022;56: 546-572.
https://doi.org/10.33594/000000575 |
| 44 | Riede UN, Schaefer HE, Wehner H: Allgemeine und spezielle Pathologie, 2. Auflage, Georg Thieme Verlag Stuttgart, New York, 1989, ISBN 3-13-683302-3.
|
| 45 | Junqueira LC, Carneiro J: Basic Histology, 1st edition, Lange Medical Publications, Los Altos, California, USA, 1971.
|
| 46 | Junqueira LC, Carneiro J: Basic Histology, 10th edition, Lange Medical Books McGraw-Hill, 2003, ISBN 0-07-121565-4.
|
| 47 | Chan FL, Inoue S, Leblond CP: The basement membranes of cryofixed or aldehyde-fixed, freeze-substituted tissues are composed of a lamina densa and do not contain a lamina lucida. Cell Tissue Res 1993;273:41-52.
https://doi.org/10.1007/BF00304610 |
| 48 | O'Rahilly R, Müller F: Developmental stages in human embryos: including a revision of Streeter's "horizons" and a survey of the Carnegie Collection, Washington, DC, Carnegie Institution of Washington Publication, 1987.
|
| 49 | Earle WR, Schilling EL, Stark TH, Straus NP, Brown MF, Shelton E: Production of malignancy in vitro. IV. The mouse fibroblast cultures and changes seen in the living cells. J Natl Cancer Inst 1942;4:165-212.
|
| 50 | Sanford KK, Eearle WR, Shelton E, Schilling EL, Duchesne EM, Likely GD, Becker MM: Production of malignancy in vitro. XII. Further transformations of mouse fibroblasts to sarcomatous cells. J Natl Cancer Inst 1950;11:351-375.
|
| 51 | Sanford KK, Likley GD, Earle WR: The development of variations in transplantability and morphology within a clone of mouse fibroblasts transformed to sarcoma-producing cells in vitro. J Natl Cancer Inst 1954;15:215-237.
|
| 52 | Sanford KK, Merwin RM, Hobbs GL, Fioramonti MC, Eearle WR: Studies on the difference in sarcoma-producing capacity of two lines of mouse cells derived in vitro from one cell. J Natl Cancer Inst 1958;20:121-145.
|
| 53 | Sanford KK, Dunn TB, Westfall BB, Covalesky AB, Dpress LT, EarleE WR: Sarcomatous change and maintenance of differentiation in long-term cultures of mouse mammary carcinoma. J Natl Cancer Inst 1961;26:1139-1183.
|
| 54 | Sanford KK, Likley GD, Bryan WR, Earle WR: The infection of cells in tissue culture with Rous sarcoma virus. J Natl Cancer Inst 1952;2:1317-1343.
|
| 55 | Stadler M, Pudelko K, Biermeier A, Walterskirchen N, Gaigneaux A, Weindorfer C, Harrer N, Klett H, Hengstschläger M, Schüler J, Sommergruber W, Oehler R, Bergmann M, Letellier E, Dolznig H: Stromal fibroblasts shape the myeloid phenotype in normal colon and colorectal cancer and induce CD163 and CCL2 expression in macrophages. Cancer Lett 2021;520:184-200.
https://doi.org/10.1016/j.canlet.2021.07.006 |
| 56 | Scott DB, Pakoskey AM, Sanford KK: Analysis of enzymatic activities of clones derived from variant cell lines transformed to malignant cells in tissue culture. J Natl Cancer Inst 1960;25:1365-1379.
|
| 57 | Sanford KK, Barker BE, Woods MW, Parshad R, Law LW: Search for "indicators" of neoplastic conversion in vitro. J Natl Cancer Inst 1967;39:705-733.
|
| 58 | Sanford KK, Jackson JL, Parshad R, Gantt RR: Evidence for an inhibiting influence of fetal bovine serum on "spontaneous" neoplastic transformation in vitro. J Natl Cancer Inst 1972;49:513-518.
|
| 59 | Gurdon JB: Factors responsible for the abnormal development of embryos obtained by nuclear transplantation in Xenopus laevis. J Embryol Exp Morphol 1960;8:327-340.
https://doi.org/10.1242/dev.8.3.327 |
| 60 | Gurdon JB: The developmental capacity of nuclei taken from differentiating endoderm cells of Xenopus laevis. J Embryol Exp Morphol 1960;8:505-526.
https://doi.org/10.1242/dev.8.4.505 |
| 61 | Gurdon JB: The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol 1962;10:622-640.
https://doi.org/10.1242/dev.10.4.622 |
| 62 | Fox CH, Caspersson T, Kudynowski J, Sanford KK, Tarone RE: Morphometric analysis of neoplastic transformation in rodent fibroblast cell lines. Cancer Res 1977;37(3):892-897.
|
| 63 | Chaudhuri S, Koprowska I, Rowinski J: Different agglutinability of fibroblasts underlying various precursor lesions of human uterine cervical carcinoma. Cancer Res 1975;35:2350-2354.
|
| 64 | Hentzer B, Kobayasi T: Adult human skin maintained in organ culture. II. The ultrastructure of the cellular compartment of connective tissue. Acta Derm Venereol 1980;60:465-476.
https://doi.org/10.2340/0001555560465476 |
| 65 | Krenning G, Zeisberg EM, Kalluri R: The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 2010;225:631-637.
https://doi.org/10.1002/jcp.22322 |
| 66 | Evans MJ, Kaufman MH: Establishment in culture of pluripotential cells from mouse embryos. Nature 1981;292:154-156.
https://doi.org/10.1038/292154a0 |
| 67 | Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663-676.
https://doi.org/10.1016/j.cell.2006.07.024 |
| 68 | Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861-872.
https://doi.org/10.1016/j.cell.2007.11.019 |
| 69 | Gordon MY, Kearney L, Hibbin JA: Effects of human marrow stromal cells on proliferation by human granulocytic (GM-CFC), erythroid (BFU-E) and mixed (Mix-CFC) colony-forming cells. Br J Haematol 1983;53:317-325.
https://doi.org/10.1111/j.1365-2141.1983.tb02026.x |
| 70 | Néchad M: Development of brown fat cells in monolayer culture. II. Ultrastructural characterization of precursors, differentiating adipocytes and their mitochondria. Exp Cell Res 1983;149:119-127.
https://doi.org/10.1016/0014-4827(83)90385-3 |
| 71 | Björntorp P: Interactions of adipocytes and their precursor cells with endothelial cells in culture. Exp Cell Res 1983;149:277-287.
https://doi.org/10.1016/0014-4827(83)90399-3 |
| 72 | Islam A: Haemopoietic stem cell: a new concept. Leuk Res 1985;9:1415-1432.
https://doi.org/10.1016/0145-2126(85)90130-4 |
| 73 | Collet AJ, Des Biens G: Evolution of mesenchymal cells in fetal rat lung. Anat Embryol (Berl) 1975;147:273-292.
https://doi.org/10.1007/BF00315076 |
| 74 | Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG: Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341-350 .
https://doi.org/10.1172/JCI0215518 |
| 75 | Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A: Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1994;1:71-81.
https://doi.org/10.1007/BF03403533 |
| 76 | Ishii G, Sangai T, Sugiyama K, Ito T, Hasebe T, Endoh Y, Magae J, Ochiai A: In vivo characterization of bone marrow-derived fibroblasts recruited into fibrotic lesions. Stem Cells 2005;23:699-706.
https://doi.org/10.1634/stemcells.2004-0183 |
| 77 | Wahlsten A, Rütsche D, Nanni M, Giampietro C, Biedermann T, Reichmann E, Mazza E: Mechanical stimulation induces rapid fibroblast proliferation and accelerates the early maturation of human skin substitutes. Biomaterials 2021;273:120779.
https://doi.org/10.1016/j.biomaterials.2021.120779 |
| 78 | Carrel A, Ebeling AH: Action of serum on fibroblasts in vitro. J Exp Med 1923;37:759-765.
https://doi.org/10.1084/jem.37.6.759 |
| 79 | Carrel A, Ebeling AH: Action on fibroblasts of extracts of homologous and heterologous tissues. J Exp Med 1923;38:499-511.
https://doi.org/10.1084/jem.38.5.499 |
| 80 | Parker RC: The races that constitute the group of common fibroblasts: I. the effect of clood plasma. J Exp Med 1932;55:713-734.
https://doi.org/10.1084/jem.55.5.713 |
| 81 | Rifkind RA, Danon D, Marks PA: Alterations in polyribosomes during erythroid cell maturation. J Cell Biol 1964;22:599-611.
https://doi.org/10.1083/jcb.22.3.599 |
| 82 | Cividalli L, Danon D: Vivo effects on maturation time of rabbit reticulocytes in the circulation. Br J Haematol 1970;19:243-250.
https://doi.org/10.1111/j.1365-2141.1970.tb01620.x |
| 83 | Appels R, Wells JR: Synthesis and turnover of DNA-bound histone during maturation of avian red blood cells. J Mol Biol 1972;70:425-434.
https://doi.org/10.1016/0022-2836(72)90550-5 |
| 84 | Fiorino AS, Diehl AM, Lin HZ, Lemischka IR, Reid LM: Maturation-dependent gene expression in a conditionally transformed liver progenitor cell line. In vitro Cell Dev Biol Anim 1998;34:247-258.
https://doi.org/10.1007/s11626-998-0131-9 |
| 85 | Lee RD, Munro SA, Knutson TP, LaRue RS, Heltemes-Harris LM, Farrar MA: Single-cell analysis identifies dynamic gene expression networks that govern B cell development and transformation. Nat Commun 2021;12:6843.
https://doi.org/10.1038/s41467-021-27232-5 |
| 86 | Wessells NK, Spooner BS, Ash JF, Bradley MO, Luduena MA, Taylor EL, Wrenn JT, Yamada K: Microfilaments in cellular and developmental processes. Science 1971;171:135-143.
https://doi.org/10.1126/science.171.3967.135 |
| 87 | Schor SL, Schor AM, Durning P, Rushton G: Skin fibroblasts obtained from cancer patients display foetal-like migratory behaviour on collagen gels. J Cell Sci 1985;73:235-244.
https://doi.org/10.1242/jcs.73.1.235 |
| 88 | Farooqui R, Fenteany G: Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell-sheet movement. J Cell Sci 2005;118:51-63.
https://doi.org/10.1242/jcs.01577 |
| 89 | Ladoux B, Nicolas A: Physically based principles of cell adhesion mechanosensitivity in tissues. Rep Prog Phys 2012;75:116601.
https://doi.org/10.1088/0034-4885/75/11/116601 |
| 90 | Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG: Identification and characterization of a fibroblast marker: FSP1 J Cell Biol 1995;130;393-405.
https://doi.org/10.1083/jcb.130.2.393 |
| 91 | Okada H, Danoff TM, Kalluri R, Neilson EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997;273:F563-574 .
https://doi.org/10.1152/ajprenal.1997.273.4.F563 |
| 92 | Fendt BM, Hirschmann A, Bruns M, Camarillo-Retamosa E, Ospelt C, Vogetseder A: Protein atlas of fibroblast specific protein 1 (FSP1)/S100A4. Research Square 2022;28:1391-1401.
https://doi.org/10.21203/rs.3.rs-1579878/v1 |
| 93 | Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG: Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341-350.
https://doi.org/10.1172/JCI0215518 |
| 94 | Prockop DJ: Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997;276:71-74.
https://doi.org/10.1126/science.276.5309.71 |
| 95 | Pallarès ME, Pi-Jaumà I, Fortunato IC, Grazu V, Gómez-González M, Roca-Cusachs P, de la Fuente JM, Alert R, Sunyer R, Casademunt J, Trepat X: Stiffness-dependent active wetting enables optimal collective cell durotaxis. Nat Physics 2022;19:279-289.
https://doi.org/10.1101/2022.07.24.501310 |
| 96 | Bray D: Axonal growth in response to experimentally applied mechanical tension. Dev Biol 1984;102:379-389.
https://doi.org/10.1016/0012-1606(84)90202-1 |
| 97 | Espina JA, Marchant CL, Barriga EH: Durotaxis: the mechanical control of directed cell migration. FEBS J 2022;289:2736-2754.
https://doi.org/10.1111/febs.15862 |
| 98 | Pathak A, Kumar S: Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc Natl Acad Sci USA 2012;109:10334-10339.
https://doi.org/10.1073/pnas.1118073109 |
| 99 | Nasrollahi S, Pathak A: Topographic confinement of epithelial clusters induces epithelial-to-mesenchymal transition in compliant matrices. Sci Rep 2016;6:18831.
https://doi.org/10.1038/srep18831 |
| 100 | Junqeira LC, Careiro J: Histologie, Springer Verlag, Berlin, 1986.
|
| 101 | Leonhart H: Histologie, Zytologie und Mikroanatomie des Menschen. Thieme Verlag, Stuttgart, 1990, ISBN 10: 3133715089.
|
| 102 | Klima J: Cytologie. Eine Einführung für Studierende der Naturwissenschaften und Medizin. Gustav Fischer Verlag, Stuttgart, 1967.
|
| 103 | Kaboth W, Begemann H: Blut, Physiologie des Menschen, Band 5, Urban & Schwarzenberg München, 1971.
|
| 104 | Leonhart H: Histologie, Zytologie und Mikroanatomie des Menschen. Thieme Verlag, Stuttgart, New York, 1985, ISBN 10: 3133715070
|
| 105 | Finch CE: Longevity, senescence and the genome. University of Chicago Press, Chicago and London, 1990, ISBN 0-226-24889-5.
|
| 106 | Miettinen PJ, Ebner R, Lopez AR, Derynck R: TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 1994;127:2021-2036.
https://doi.org/10.1083/jcb.127.6.2021 |
| 107 | Krug EL, Mjaatvedt CH, Markwald RR: Extracellular matrix from embryonic myocardium elicits an early morphogenetic event in cardiac endothelial differentiation. Dev Biol 1987;120:348-355.
https://doi.org/10.1016/0012-1606(87)90237-5 |
| 108 | Bae SN, Arand G, Azzam H, Pavasant P, Torri J, Frandsen TL, Thompson EW: Molecular and cellular analysis of basement membrane invasion by human breast cancer cells in Matrigel-based in vitro assays. Breast Cancer Res Treat 1993;24:241-255.
https://doi.org/10.1007/BF01833264 |
| 109 | Heikinheimo K, Happonen RP, Miettinen PJ, Ritvos O: Transforming growth factor beta 2 in epithelial differentiation of developing teeth and odontogenic tumors. J Clin Invest 1993; 91:1019-1027.
https://doi.org/10.1172/JCI116258 |
| 110 | Pulyaeva H, Bueno J, Polette M, Birembaut P, Sato H, Seiki M, Thompson EW: MT1-MMP correlates with MMP-2 activation potential seen after epithelial to mesenchymal transition in human breast carcinoma cells. Clin Exp Metastasis 1997;15:111-120.
https://doi.org/10.1023/A:1018444609098 |
| 111 | Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Müller A, Tuschl G, Mueller SO, Dooley S: Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology 2009;49:2031-2043.
https://doi.org/10.1002/hep.22880 |
| 112 | Buckley ST, Medina C, Davies AM, Ehrhardt C: Cytoskeletal re-arrangement in TGF-β1-induced alveolar epithelial-mesenchymal transition studied by atomic force microscopy and high-content analysis. Nanomedicine 2012;8:355-364.
https://doi.org/10.1016/j.nano.2011.06.021 |
| 113 | Brown AC, Fiore VF, Sulchek TA, Barker TH: Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions. J Pathol 2013;229:25-35.
https://doi.org/10.1002/path.4114 |
| 114 | Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS: Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell 2012;23:781-791.
https://doi.org/10.1091/mbc.e11-06-0537 |
| 115 | Birchmeier C, Birchmeier W, Brand-Saberi B: Epithelial-mesenchymal transitions in cancer progression. Acta Anat (Basel) 1996;156:217-226.
https://doi.org/10.1159/000147848 |
| 116 | Yao J, Cui Q, Fan W, Ma Y, Chen Y, Liu T, Zhang X, Xi Y, Wang C, Peng L, Luo Y, Lin A, Guo W, Lin L, Lin Y, Tan W, Lin D, Wu C, Wang J: Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer. Nat Commun 2020;11:3715.
https://doi.org/10.1038/s41467-020-17492-y |
| 117 | Michael H, Ulbright TM, Brodhecker CA: The pluripotential nature of the mesenchyme-like component of yolk sac tumor. Arch Pathol Lab Med 1989;113:1115-1119.
|
| 118 | Martins-Neves SR, Paiva-Oliveira DI, Wijers-Koster PM, Abrunhosa AJ, Fontes-Ribeiro C, Bovée JV, Cleton-Jansen AM, Gomes CM: Chemotherapy induces stemness in osteosarcoma cells through activation of Wnt/β-catenin signaling. Cancer Lett 2016;370:286-295.
https://doi.org/10.1016/j.canlet.2015.11.013 |
| 119 | Larocca LM, Teofili L, Maggiano N, Piantelli M, Ranelletti FO, Leone G: Quercetin and the growth of leukemic progenitors. Leuk Lymphoma 1996;23:49-53.
https://doi.org/10.3109/10428199609054801 |
| 120 | Klein F, Feldhahn N, Mooster JL, Sprangers M, Hofmann WK, Wernet P, Wartenberg M, Müschen M: Tracing the pre-B to immature B cell transition in human leukemia cells reveals a coordinated sequence of primary and secondary IGK gene rearrangement, IGK deletion, and IGL gene rearrangement. J Immunol 2005;174:367-375.
https://doi.org/10.4049/jimmunol.174.1.367 |
| 121 | Mehta A, Mann M, Zhao JL, Marinov GK, Majumdar D, Garcia-Flores Y, Du X, Erikci E, Chowdhury K, Baltimore D: The microRNA-212/132 cluster regulates B cell development by targeting Sox4. J Exp Med 2015;212:1679-1692.
https://doi.org/10.1084/jem.20150489 |
| 122 | Smeenk L, Fischer M, Jurado S, Jaritz M, Azaryan A, Werner B, Roth M, Zuber J, Stanulla M, den Boer ML, Mullighan CG, Strehl S, Busslinger M: Molecular role of the PAX5-ETV6 oncoprotein in promoting B-cell acute lymphoblastic leukemia. EMBO J 2017;36:718-735.
https://doi.org/10.15252/embj.201695495 |
| 123 | Mack R, Zhang L, Breslin Sj P, Zhang J: The Fetal-to-Adult Hematopoietic Stem Cell Transition and its Role in Childhood Hematopoietic Malignancies. Stem Cell Rev Rep 2021;17:2059-2080.
https://doi.org/10.1007/s12015-021-10230-x |
| 124 | Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, Stoppacciaro A, Tiveron C, Tatangelo L, Giovarelli M, Gaetano C, Ruco L, Hoffman ES, Hayday AC, Lendahl U, Frati L, Gulino A, Screpanti I: Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J 2000;19:337-3348.
https://doi.org/10.1093/emboj/19.13.3337 |
| 125 | Tooze RM: A replicative self-renewal model for long-lived plasma cells: questioning irreversible cell cycle exit. Front Immunol 2013;4460.
https://doi.org/10.3389/fimmu.2013.00460 |
| 126 | Nobari ST, Nojadeh JN, Talebi M: B-cell maturation antigen targeting strategies in multiple myeloma treatment, advantages and disadvantages. J Transl Med 2022;20:82.
https://doi.org/10.1186/s12967-022-03285-y |
| 127 | Elias MC, Tozer KR, Silber JR, Mikheeva S, Deng M, Morrison RS, Manning TC, Silbergeld DL, Glackin CA, Reh TA, Rostomily RC: TWIST is expressed in human gliomas and promotes invasion. Neoplasia 2005;7:824-837.
https://doi.org/10.1593/neo.04352 |
| 128 | Priester M, Copanaki E, Vafaizadeh V, Hensel S, Bernreuther C, Glatzel M, Seifert V, Groner B, Kögel D, Weissenberger J: STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro Oncol 2013;15:840-852.
https://doi.org/10.1093/neuonc/not025 |
| 129 | Tanabe K, Nakamura M, Narita M, Takahashi K, Yamanaka S: Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc Natl Acad Sci USA 2013;110:12172-12179.
https://doi.org/10.1073/pnas.1310291110 |
| 130 | Hay ED: Interaction of embryonic surface and cytoskeleton with extracellular matrix. Am J Anat 1982;165:1-12.
https://doi.org/10.1002/aja.1001650102 |
| 131 | Hay ED: Theory for epithelial-mesenchymal transformation based on the "fixed cortex" cell motility model. Cell Motil Cytoskeleton 1989;14:455-457.
https://doi.org/10.1002/cm.970140403 |
| 132 | Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S: Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res 2009;11:R46.
https://doi.org/10.1186/bcr2333 |
| 133 | Mitchel JA, Das A, O'Sullivan MJ, Stancil IT, DeCamp SJ, Koehler S, Ocaña OH, Butler JP, Fredberg JJ, Nieto MA, Bi D, Park JA: In primary airway epithelial cells, the unjamming transition is distinct from the epithelial-to-mesenchymal transition. Nat Commun 2020;11:5053.
https://doi.org/10.1038/s41467-020-18841-7 |
| 134 | De Marzio M, Kılıç A, Maiorino E, Mitchel JA, Mwase C, O'Sullivan MJ, McGill M, Chase R, Fredberg JJ, Park JA, Glass K, Weiss ST: Genomic signatures of the unjamming transition in compressed human bronchial epithelial cells. Sci Adv 2021;7:eabf1088.
https://doi.org/10.1126/sciadv.abf1088 |
| 135 | Haeger A, Krause M, Wolf K, Friedl P: Cell jamming: Collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim Biophys Acta 2014;1840:2386-2395.
https://doi.org/10.1016/j.bbagen.2014.03.020 |
| 136 | Park JA, Kim JH, Bi D, Mitchel JA, Qazvini NT, Tantisira K, Park CY, McGill M, Kim SH, Gweon B, Notbohm J, Steward R Jr, Burger S, Randell SH, Kho AT, Tambe DT, Hardin C, Shore SA, Israel E, Weitz DA, Tschumperlin DJ, Henske EP, Weiss ST, Manning ML, Butler JP, Drazen JM, Fredberg JJ (2015) Unjamming and cell shape in the asthmatic airway epithelium. Nat Mater 2015;14:1040-1048.
https://doi.org/10.1038/nmat4357 |
| 137 | Nandi T, Yadav A, Ainavarapu SRK: Experimental comparison of energy landscape features of ubiquitin family proteins. Proteins 2020;88:449-461.
https://doi.org/10.1002/prot.25822 |
| 138 | Meluh PB, Koshland D: Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol Biol Cell 1995;6:793-807.
https://doi.org/10.1091/mbc.6.7.793 |
| 139 | Mahajan R, Delphin C, Guan T, Gerace L, Melchior F: A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997;88:97-107.
https://doi.org/10.1016/S0092-8674(00)81862-0 |
| 140 | Boddy MN, Howe K, Etkin LD, Solomon E, Freemont PS: PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 1996;13:971-982.
|
| 141 | Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, Solomon E, de Thé H, Hay RT, Freemont PS: SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localization. J Cell Sci 1999;112:381-393.
https://doi.org/10.1242/jcs.112.3.381 |
| 142 | Sternsdorf T, Jensen K, Reich B, Will H: The nuclear dot protein sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J Biol Chem 1999;274:12555-12566.
https://doi.org/10.1074/jbc.274.18.12555 |
| 143 | Kerscher O: SUMO junction-what's your function? New insights through SUMO-interacting motifs. EMBO Rep 2007;8:550-555.
https://doi.org/10.1038/sj.embor.7400980 |
| 144 | Celen AB, Sahin U: Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts. FEBS J 2020;287:3110-3140.
https://doi.org/10.1111/febs.15319 |
| 145 | Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G: The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J 1977;16:5509-5519.
https://doi.org/10.1093/emboj/16.18.5509 |
| 146 | Desterro JM, Rodriguez MS, Kemp GD, Hay RT: Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1, J Biol Chem 1999;274:10618-10624.
https://doi.org/10.1074/jbc.274.15.10618 |
| 147 | Geiss-Friedlander R, Melchior F (2007) Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol;8:947-956.
https://doi.org/10.1038/nrm2293 |
| 148 | Yahiro K, Tsutsuki H, Ogura K, Nagasawa S, Moss J, Noda M: DAP1, a negative regulator of autophagy, controls SubAB-mediated apoptosis and autophagy. Infect Immun 2014;82:4899-4908.
https://doi.org/10.1128/IAI.02213-14 |
| 149 | Lorente M, García-Casas A, Salvador N, Martínez-López A, Gabicagogeascoa E, Velasco G, López-Palomar L, Castillo-Lluva S: Inhibiting SUMO1-mediated SUMOylation induces autophagy-mediated cancer cell death and reduces tumour cell invasion via RAC1. J Cell Sci 2019;132(20):jcs234120.
https://doi.org/10.1242/jcs.234120 |
| 150 | Agbor TA, Cheong A, Comerford KM, Scholz CC, Bruning U, Clarke A, Cummins EP, Cagney G, Taylor CT: Small ubiquitin-related modifier (SUMO)-1 promotes glycolysis in hypoxia. J Biol Chem 2011;286:4718-4726.
https://doi.org/10.1074/jbc.M110.115931 |
| 151 | Su X, Wan Y, Xie L, Lin X, Zhao H, Ju X, Fang A: Expression of SUMO1P3 Compared with SUMO1 is an Independent Predictor of Patient Outcome in Lung Adenocarcinoma. Med Sci Monit 2019;25:6691-6701.
https://doi.org/10.12659/MSM.916887 |
| 152 | Zhang Y, Li Y, Han L, Zhang P, Sun S: SUMO1P3 is associated clinical progression and facilitates cell migration and invasion through regulating miR-136 in non-small cell lung cancer. Biomed Pharmacother 2019;113:108686
https://doi.org/10.1016/j.biopha.2019.108686 |
| 153 | Yang W, Wang L, Roehn G, Pearlstein RD, Ali-Osman F, Pan H, Goldbrunner R, Krantz M, Harms C, Paschen W: Small ubiquitin-like modifier 1-3 conjugation [corrected] is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci 2013;104:70-77.
https://doi.org/10.1111/cas.12047 |
| 154 | Ke J, Yang Y, Che Q, Jiang F, Wang H, Chen Z, Zhu M, Tong H, Zhang H, Yan X, Wang X, Wang F, Liu Y, Dai C, Wan X: Prostaglandin E2 (PGE2) promotes proliferation and invasion by enhancing SUMO-1 activity via EP4 receptor in endometrial cancer. Tumour Biol 2016;37:12203-12211.
https://doi.org/10.1007/s13277-016-5087-x |
| 155 | Oliveira Alves MG, da Mota Delgado A, Balducci I, Carvalho YR, Cavalcante AS, Almeida JD: Study of MDM2 and SUMO-1 expression in actinic cheilitis and lip cancer. Arch Dermatol Res 2014;306:837-841.
https://doi.org/10.1007/s00403-014-1500-8 |
| 156 | Liu J, Tao X, Zhang J, Wang P, Sha M, Ma Y, Geng X, Feng L, Shen Y, Yu Y, Wang S, Fang S, Shen Y: Small ubiquitin-related modifier 1 is involved in hepatocellular carcinoma progression via mediating p65 nuclear translocation. Oncotarget 2016;7:22206-22218.
https://doi.org/10.18632/oncotarget.8066 |
| 157 | Yu H, Bai Y, Xu C, He X, Liu Q, Ma D, A Y: The tissue expression levels of SUMO1P 3 may be a reliable prognostic biomarker to predict the clinical outcomes in patients with HCC. Medicine (Baltimore) 2020;99:e21291.
https://doi.org/10.1097/MD.0000000000021291 |
| 158 | Mei D, Song H, Wang K, Lou Y, Sun W, Liu Z, Ding X, Guo J: Up-regulation of SUMO1 pseudogene 3 (SUMO1P3) in gastric cancer and its clinical association. Med Oncol 2013;30:709.
https://doi.org/10.1007/s12032-013-0709-2 |
| 159 | Duan Y, Du Y, Mu Y, Gu Z, Wang C: Prognostic value, immune signature and molecular mechanisms of the SUMO family in pancreatic adenocarcinoma. Front Mol Biosci 2022;9:1096679 .
https://doi.org/10.3389/fmolb.2022.1096679 |
| 160 | El Motiam A, Vidal S, Seoane R, Bouzaher YH, González-Santamaría J, Rivas C: SUMO and Cytoplasmic RNA Viruses: From Enemies to Best Friends. Adv Exp Med Biol 2020;1233:263-277.
https://doi.org/10.1007/978-3-030-38266-7_11 |
| 161 | Chelbi-Alix MK, Thibault P: Crosstalk Between SUMO and Ubiquitin-Like Proteins: Implication for Antiviral Defense. Front Cell Dev Biol 2020;9:671067.
https://doi.org/10.3389/fcell.2021.671067 |
| 162 | Rangasamy D, Woytek K, Khan SA, Wilson VG: SUMO-1 modification of bovine papillomavirus E1 protein is required for intranuclear accumulation. J Biol Chem 2000;275:37999-38004.
https://doi.org/10.1074/jbc.M007777200 |
| 163 | Heider JA, Yu Y, Shenk T, Alwine JC: Characterization of a human cytomegalovirus with phosphorylation site mutations in the immediate-early 2 protein. J Virol 2002;76:928-932.
https://doi.org/10.1128/JVI.76.2.928-932.2002 |
| 164 | Chang LK, Lee YH, Cheng TS, Hong YR, Lu PJ, Wang JJ, Wang WH, Kuo CW, Li SS, Liu ST (2004) Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J Biol Chem 2004;279:38803-38812.
https://doi.org/10.1074/jbc.M405470200 |
| 165 | Adamson AL: Effects of SUMO-1 upon Epstein-Barr virus BZLF1 function and BMRF1 expression. Biochem Biophys Res Commun 2005;336:22-28.
https://doi.org/10.1016/j.bbrc.2005.08.036 |
| 166 | Tan M, Zhang D, Zhang E, Xu D, Liu Z, Qiu J, Fan Y, Shen B (2017) SENP2 suppresses epithelial-mesenchymal transition of bladder cancer cells through deSUMOylation of TGF-βRI. Mol Carcinog 2017;56:2332-2341.
https://doi.org/10.1002/mc.22687 |
| 167 | Khodzhigorova A, Distler A, Lang V, Dees C, Schneider H, Beyer C, Gelse K, Distler O, Schett G, Distler JH: Inhibition of sumoylation prevents experimental fibrosis. Ann Rheum Dis 2012;71:1904-1908.
https://doi.org/10.1136/annrheumdis-2012-201746 |
| 168 | Ungefroren H: TGF-β Signaling in Cancer: Control by Negative Regulators and Crosstalk with Proinflammatory and Fibrogenic Pathways. Cancers (Basel) 2019;11:384.
https://doi.org/10.3390/cancers11030384 |
| 169 | Lin X, Wang Y, Jiang Y, Xu M, Pang Q, Sun J, Yu Y, Shen Z, Lei R, Xu J: Sumoylation enhances the activity of the TGF-β/SMAD and HIF-1 signaling pathways in keloids. Life Sci 2020;255:117859.
https://doi.org/10.1016/j.lfs.2020.117859 |
| 170 | Wang X, Liu T, Huang Y, Dai Y, Lin H: Regulation of transforming growth factor-β signalling by SUMOylation and its role in fibrosis. Open Biol 2021;11:210043.
https://doi.org/10.1098/rsob.210043 |
| 171 | Fischer H, Salahshor S, Stenling R, Björk J, Lindmark G, Iselius L, Rubio C, Lindblom A: COL11A1 in FAP polyps and in sporadic colorectal tumors. BMC Cancer 2001;1:17.
https://doi.org/10.1186/1471-2407-1-17 |
| 172 | Kikuchi Y, Kashima TG, Nishiyama T, Shimazu K, Morishita Y, Shimazaki M, Kii I, Horie H, Nagai H, Kudo A, Fukayama M: Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J Histochem Cytochem 2008;56:753-764.
https://doi.org/10.1369/jhc.2008.951061 |
| 173 | Zhang D, Zhu H, Harpaz N: Overexpression of α1 chain of type XI collagen (COL11A1) aids in the diagnosis of invasive carcinoma in endoscopically removed malignant colorectal polyps. Pathol Res Pract 2016;212:545-548.
https://doi.org/10.1016/j.prp.2016.03.005 |
| 174 | Becker WR, Nevins SA, Chen DC, Chiu R, Horning AM, Guha TK, Laquindanum R, Mills M, Chaib H, Ladabaum U, Longacre T, Shen J, Esplin ED, Kundaje A, Ford JM, Curtis C, Snyder MP, Greenleaf WJ: Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet 2022;54:985-995.
https://doi.org/10.1038/s41588-022-01088-x |
| 175 | Kawamura T, Yamamoto M, Suzuki K, Suzuki Y, Kamishima M, Sakata M, Kurachi K, Setoh M, Konno H, Takeuchi H: Tenascin-C Produced by Intestinal Myofibroblasts Promotes Colitis-associated Cancer Development Through Angiogenesis. Inflamm Bowel Dis 2019;25:732-741.
https://doi.org/10.1093/ibd/izy368 |
| 176 | Leppänen J, Bogdanoff S, Lehenkari PP, Saarnio J, Kauppila JH, Karttunen TJ, Huhta H, Helminen O: Tenascin-C and fibronectin in normal esophageal mucosa, Barrett's esophagus, dysplasia and adenocarcinoma. Oncotarget 2017;8:66865-66877.
https://doi.org/10.18632/oncotarget.19196 |
| 177 | Qin Y, Wang F, Ni H, Liu Y, Yin Y, Zhou X, Gao G, Li Q, Qi X, Li J: Cancer-associated fibroblasts in gastric cancer affect malignant progression via the CXCL12-CXCR4 axis. J Cancer 2021;12:3011-3023.
https://doi.org/10.7150/jca.49707 |
| 178 | Akrish SJ, Rachmiel A, Sabo E, Vered M, Ben-Izhak O: Cancer-associated fibroblasts are an infrequent finding in the microenvironment of proliferative verrucous leukoplakia-associated squamous cell carcinoma. J Oral Pathol Med 2017;46:353-358.
https://doi.org/10.1111/jop.12503 |
| 179 | Fang Y, Chen M, Li G, Yang Y, He P, Chen J, Cheng L, Wu H: Cancer-associated fibroblast-like fibroblasts in vocal fold leukoplakia suppress CD8+T cell functions by inducing IL-6 autocrine loop and interacting with Th17 cells. Cancer Lett 2022;546:215839.
https://doi.org/10.1016/j.canlet.2022.215839 |
| 180 | Lyu L, Jiang Y, Ma W, Li H, Liu X, Li L, Shen A, Yu Y, Jiang S, Li H, Zhou P, Yin S: Single-cell sequencing of PIT1-positive pituitary adenoma highlights the pro-tumour microenvironment mediated by IFN-γ-induced tumour-associated fibroblasts remodeling. Br J Cancer 2023;128:1117-1133.
https://doi.org/10.1038/s41416-022-02126-5 |
| 181 | Tancred TM, Belch AR, Reiman T, Pilarski LM, Kirshner J: Altered expression of fibronectin and collagens I and IV in multiple myeloma and monoclonal gammopathy of undetermined significance. J Histochem Cytochem 2009;57:239-247.
https://doi.org/10.1369/jhc.2008.952200 |
| 182 | Asimakopoulos F, Hope C, Johnson MG, Pagenkopf A, Gromek K, Nagel B: Extracellular matrix and the myeloid-in-myeloma compartment: balancing tolerogenic and immunogenic inflammation in the myeloma niche. J Leukoc Biol 2017;102:265-275.
https://doi.org/10.1189/jlb.3MR1116-468R |
| 183 | Ciavarella S, Laurenzana A, De Summa S, Pilato B, Chillà A, Lacalamita R, Minoia C, Margheri F, Iacobazzi A, Rana A, Merchionne F, Fibbi G, Del Rosso M, Guarini A, Tommasi S, Serratì S: u-PAR expression in cancer associated fibroblast: new acquisitions in multiple myeloma progression. BMC Cancer 2017;17:215.
https://doi.org/10.1186/s12885-017-3183-y |
| 184 | Ni WD, Yang ZT, Cui CA, Cui Y, Fang LY, Xuan YH: Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem Biophys Res Commun 2017;486:607-612.
https://doi.org/10.1016/j.bbrc.2017.03.021 |
| 185 | Ferguson JE, Schor AM, Howell A, Ferguson MW: Tenascin distribution in the normal human breast is altered during the menstrual cycle and in carcinoma. Differentiation 1990;42:199-207.
https://doi.org/10.1111/j.1432-0436.1990.tb00762.x |
| 186 | Barth PJ, Ebrahimsade S, Ramaswamy A, Moll R: CD34+ fibrocytes in invasive ductal carcinoma, ductal carcinoma in situ, and benign breast lesions. Virchows Arch 2002;440:298-303.
https://doi.org/10.1007/s004280100530 |
| 187 | Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, Schnitt S, Sellers WR, Polyak K: Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004;6:17-32.
https://doi.org/10.1016/j.ccr.2004.06.010 |
| 188 | Hu M, Yao J, Cai L, Bachman KE, van den Brûle F, Velculescu V, Polyak K: Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet 2005;37:899-905.
https://doi.org/10.1038/ng1596 |
| 189 | Visscher DW, Pankratz VS, Santisteban M, Reynolds C, Ristimäki A, Vierkant RA, Lingle WL, Frost MH, Hartmann LC: Association between cyclooxygenase-2 expression in atypical hyperplasia and risk of breast cancer. J Natl Cancer Inst 2008;100:421-427.
https://doi.org/10.1093/jnci/djn036 |
| 190 | Shaker H, Bundred NJ, Albadry H, Nicholson S, Castle J, Lumsden LJ, Pritchard S, Landberg G, Kirwan CC: PO-21 - Stromal fibroblasts in preinvasive breast cancer (ductal carcinoma in situ, DCIS) demonstrate a cancer-like procoagulant phenotypic switch that may facilitate invasion. Thromb Res 2016;140(Suppl1):S184 .
https://doi.org/10.1016/S0049-3848(16)30154-2 |
| 191 | Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, McCaffrey EF, Greenwald NF, Rivero-Gutiérrez B, Strand SH, Varma S, Kong A, Keren L, Srivastava S, Zhu C, Khair Z, Veis DJ, Deschryver K, Vennam S, Maley C, Hwang ES, Marks JR, Bendall SC, Colditz GA, West RB, Angelo M: Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 2022;185:299-310.e18.
https://doi.org/10.1016/j.cell.2021.12.023 |
| 192 | Matsuda Y, Ye J, Yamakawa K, Mukai Y, Azuma K, Wu L, Masutomi K, Yamashita T, Daigo Y, Miyagi Y, Yokose T, Oshima T, Ito H, Morinaga S, Kishida T, Minamoto T, Kojima M, Kaneko S, Haba R, Kontani K, Kanaji N, Okano K, Muto-Ishizuka M, Yokohira M, Saoo K, Imaida K, Suizu F: Association of longer telomere length in cancer cells and cancer-associated fibroblasts with worse prognosis. J Natl Cancer Inst 2023;115:208-218.
https://doi.org/10.1093/jnci/djac226 |
| 193 | Takeuchi Y, Gotoh N: Inflammatory cytokine-enriched microenvironment plays key roles in the development of breast cancers. Cancer Sci 2023;114:1792-1799.
https://doi.org/10.1111/cas.15734 |
| 194 | Tarnowski M, Grymula K, Liu R, Tarnowska J, Drukala J, Ratajczak J, Mitchell RA, Ratajczak MZ, Kucia M: Macrophage migration inhibitory factor is secreted by rhabdomyosarcoma cells, modulates tumor metastasis by binding to CXCR4 and CXCR7 receptors and inhibits recruitment of cancer-associated fibroblasts. Mol Cancer 2010;Res 8:1328-1343.
https://doi.org/10.1158/1541-7786.MCR-10-0288 |
| 195 | Wang JW, Wu XF, Gu XJ, Jiang XH: Exosomal miR-1228 From Cancer-Associated Fibroblasts Promotes Cell Migration and Invasion of Osteosarcoma by Directly Targeting SCAI. Oncol Res 2019;27:979-986
https://doi.org/10.3727/096504018X15336368805108 |
| 196 | Huang X, Wang L, Guo H, Zhang W, Shao Z: Single-cell transcriptomics reveals the regulative roles of cancer associated fibroblasts in tumor immune microenvironment of recurrent osteosarcoma. Theranostics 2022;12:5877-5887.
https://doi.org/10.7150/thno.73714 |
| 197 | Frassanito MA, Rao L, Moschetta M, Ria R, Di Marzo L, De Luisi A, Racanelli V, Catacchio I, Berardi S, Basile A, Menu E, Ruggieri S, Nico B, Ribatti D, Fumarulo R, Dammacco F, Vanderkerken K, Vacca A: Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia 2014;28:904-916
https://doi.org/10.1038/leu.2013.254 |
| 198 | Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, Kim YJ, Adam J, Lichter P, Solary E, Berchem G, Moussay E: Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015;126:1106-1117 .
https://doi.org/10.1182/blood-2014-12-618025 |
| 199 | Frassanito MA, De Veirman K, Desantis V, Di Marzo L, Vergara D, Ruggieri S, Annese T, Nico B, Menu E, Catacchio I, Ria R, Racanelli V, Maffia M, Angelucci E, Derudas D, Fumarulo R, Dammacco F, Ribatti D, Vanderkerken K, Vacca A: Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2016;30:640-648.
https://doi.org/10.1038/leu.2015.289 |
| 200 | Raffaghello L, Vacca A, Pistoia V, Ribatti D: Cancer associated fibroblasts in hematological malignancies. Oncotarget 2015;6:2589-603.
https://doi.org/10.18632/oncotarget.2661 |
| 201 | Zhai Y, Zhang J, Wang H, Lu W, Liu S, Yu Y, Weng W, Ding Z, Zhu Q, Shi J: Growth differentiation factor 15 contributes to cancer-associated fibroblasts-mediated chemo-protection of AML cells. J Exp Clin Cancer Res 2016;35:147.
https://doi.org/10.1186/s13046-016-0405-0 |
| 202 | Staiger AM, Duppel J, Dengler MA, van der Kuip H, Vöhringer MC, Aulitzky WE, Rosenwald A, Ott G, Horn H: An analysis of the role of follicular lymphoma-associated fibroblasts to promote tumor cell viability following drug-induced apoptosis. Leuk Lymphoma 2017;58:1922-1930.
https://doi.org/10.1080/10428194.2016.1263841 |
| 203 | Dörsam B, Bösl T, Reiners KS, Barnert S, Schubert R, Shatnyeva O, Zigrino P, Engert A, Hansen HP, von Strandmann EP: Hodgkin Lymphoma-Derived Extracellular Vesicles Change the Secretome of Fibroblasts Toward a CAF Phenotype. Front Immunol 2018:9:1358.
https://doi.org/10.3389/fimmu.2018.01358 |
| 204 | Sakamoto A, Kunou S, Shimada K, Tsunoda M, Aoki T, Iriyama C, Tomita A, Nakamura S, Hayakawa F, Kiyoi H: Pyruvate secreted from patient-derived cancer-associated fibroblasts supports survival of primary lymphoma cells. Cancer Sci 2019;110:269-278.
https://doi.org/10.1111/cas.13873 |
| 205 | Lamaison C, Tarte K: B cell/stromal cell crosstalk in health, disease, and treatment: Follicular lymphoma as a paradigm. Immunol Rev 2021;302:273-285.
https://doi.org/10.1111/imr.12983 |
| 206 | Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, Manriquez Roman C, Schick KJ, Can I, Tapper EE, Horvei P, Adada MM, Bezerra ED, Kankeu Fonkoua LA, Ruff MW, Nevala WK, Walters DK, Parikh SA, Lin Y, Jelinek DF, Kay NE, Bergsagel PL, Kenderian SS: Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood 2022;139:3708-3721.
https://doi.org/10.1182/blood.2021012811 |
| 207 | Trylcova J, Busek P, Smetana K Jr, Balaziova E, Dvorankova B, Mifkova A, Sedo A: Effect of cancer-associated fibroblasts on the migration of glioma cells in vitro. Tumour Biol 2015;36:5873-5879.
https://doi.org/10.1007/s13277-015-3259-8 |
| 208 | Prajapati P, Lambert DW: Cancer-associated fibroblasts - Not-so-innocent bystanders in metastasis to bone? J Bone Oncol 2016;5:128-131.
https://doi.org/10.1016/j.jbo.2016.03.008 |
| 209 | Li M, Li G, Kiyokawa J, Tirmizi Z, Richardson LG, Ning J, Das S, Martuza RL, Stemmer-Rachamimov A, Rabkin SD, Wakimoto H: Characterization and oncolytic virus targeting of FAP-expressing tumor-associated pericytes in glioblastoma. Acta Neuropathol Commun 2020;8:221
https://doi.org/10.1186/s40478-020-01096-0 |
| 210 | Jain S, Rick JW, Joshi RS, Beniwal A, Spatz J, Gill S, Chang AC, Choudhary N, Nguyen AT, Sudhir S, Chalif EJ, Chen JS, Chandra A, Haddad AF, Wadhwa H, Shah SS, Choi S, Hayes JL, Wang L, Yagnik G, Costello JF, Diaz A, Heiland DH, Aghi MK: Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. J Clin Invest 2023;133:e147087.
https://doi.org/10.1172/JCI147087 |
| 211 | Yoshida GJ: Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways, J Exp Clin Cancer Res 2020;39:112.
https://doi.org/10.1186/s13046-020-01611-0 |
| 212 | Biffi G, Tuveson DA: Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev 2021;101:147-176.
https://doi.org/10.1152/physrev.00048.2019 |
| 213 | Yoon H, Tang CM, Banerjee S, Delgado AL, Yebra M, Davis J, Sicklick JK: TGF-β1-mediated transition of resident fibroblasts to cancer-associated fibroblasts promotes cancer metastasis in gastrointestinal stromal tumor. Oncogenesis 2021;10:13.
https://doi.org/10.1038/s41389-021-00302-5 |
| 214 | Mir S, Golden BDO, Griess BJ, Vengoji R, Tom E, Kosmacek EA, Oberley-Deegan RE, Talmon GA, Band V, Teoh-Fitzgerald ML: Upregulation of Nox4 induces a pro-survival Nrf2 response in cancer-associated fibroblasts that promotes tumorigenesis and metastasis, in part via Birc5 induction. Breast Cancer Res 2022;24:48.
https://doi.org/10.1186/s13058-022-01548-6 |
| 215 | Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D: NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005;97:900-907.
https://doi.org/10.1161/01.RES.0000187457.24338.3D |
| 216 | Moi P, Chan K, Asunis I, Cao A, Kan YW: Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA 1994;91:9926-9930.
https://doi.org/10.1073/pnas.91.21.9926 |
| 217 | Wu R, Roy AM, Tokumaru Y, Gandhi S, Asaoka M, Oshi M, Yan L, Ishikawa T, Takabe K: NR2F1, a Tumor Dormancy Marker, Is Expressed Predominantly in Cancer-Associated Fibroblasts and Is Associated with Suppressed Breast Cancer Cell Proliferation. Cancers (Basel) 2022;14:2962.
https://doi.org/10.3390/cancers14122962 |
| 218 | Miao W, Hu L, Scrivens PJ, Batist G: Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem 2005;280:20340-20348
https://doi.org/10.1074/jbc.M412081200 |
| 219 | Feng H, Cao B, Peng X, Wei Q: Cancer-associated fibroblasts strengthen cell proliferation and EGFR TKIs resistance through aryl hydrocarbon receptor dependent signals in non-small cell lung cancer. BMC Cancer 2022;22:764
https://doi.org/10.1186/s12885-022-09877-7 |
| 220 | Therachiyil L, Krishnankutty R, Ahmad F, Mateo JM, Uddin S, Korashy HM: Aryl Hydrocarbon Receptor Promotes Cell Growth, Stemness Like Characteristics, and Metastasis in Human Ovarian Cancer via Activation of PI3K/Akt, β-Catenin, and Epithelial to Mesenchymal Transition Pathways. Int J Mol Sci 2022;23:6395.
https://doi.org/10.3390/ijms23126395 |
| 221 | Li CH, Liu CW, Tsai CH, Peng YJ, Yang YH, Liao PL, Lee CC, Cheng YW, Kang JJ: Cytoplasmic aryl hydrocarbon receptor regulates glycogen synthase kinase 3 beta, accelerates vimentin degradation, and suppresses epithelial-mesenchymal transition in non-small cell lung cancer cells. Arch Toxicol 2017;91:2165-2178.
https://doi.org/10.1007/s00204-016-1870-0 |
| 222 | Zhu P, Yu H, Zhou K, Bai Y, Qi R, Zhang S: 3, 3'-Diindolylmethane modulates aryl hydrocarbon receptor of esophageal squamous cell carcinoma to reverse epithelial-mesenchymal transition through repressing RhoA/ROCK1-mediated COX2/PGE2 pathway. J Exp Clin Cancer Res 2020;39:113.
https://doi.org/10.1186/s13046-020-01618-7 |
| 223 | Mariner DJ, Anastasiadis P, Keilhack H, Böhmer FD, Wang J, Reynolds AB: Identification of Src phosphorylation sites in the catenin p120ctn. J Biol Chem 2001;276:28006-28013.
https://doi.org/10.1074/jbc.M102443200 |
| 224 | Shibata T, Kokubu A, Sekine S, Kanai Y, Hirohashi S: Cytoplasmic p120ctn regulates the invasive phenotypes of E-cadherin-deficient breast cancer. Am J Pathol 2004;164:2269-2278.
https://doi.org/10.1016/S0002-9440(10)63783-2 |
| 225 | Dabbs DJ, Bhargava R, Chivukula M: Lobular versus ductal breast neoplasms: the diagnostic utility of p120 catenin. Am J Surg Pathol 2007;31:427-437.
https://doi.org/10.1097/01.pas.0000213386.63160.3f |
| 226 | McCart Reed AE, Kutasovic JR, Lakhani SR, Simpson PT: Invasive lobular carcinoma of the breast: morphology, biomarkers and 'omics. Breast Cancer Res 2015;17:12.
https://doi.org/10.1186/s13058-015-0519-x |
| 227 | Belgiovine C, Chiodi I, Mondello C: Relocalization of cell adhesion molecules during neoplastic transformation of human fibroblasts. Int J Oncol 2011;39:1199-1204.
|
| 228 | Noren NK, Liu BP, Burridge K, Kreft B: p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 2000;150:567-580.
https://doi.org/10.1083/jcb.150.3.567 |
| 229 | Grosheva I, Shtutman M, Elbaum M, Bershadsky AD: p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J Cell Sci 2001;114:695-707.
https://doi.org/10.1242/jcs.114.4.695 |
| 230 | Tran NL, Nagle RB, Cress AE, Heimark RL: N-Cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion withStromal cells. Am J Pathol 1999;155:787-798.
https://doi.org/10.1016/S0002-9440(10)65177-2 |
| 231 | Fonagy A, Swiderski C, Wilson A, Bolton W, Kenyon N, Freeman JW: Cell cycle regulated expression of nucleolar antigen P120 in normal and transformed human fibroblasts. J Cell Physiol 1993;154:16-27.
https://doi.org/10.1002/jcp.1041540104 |
| 232 | Twardzik DR, Todaro GJ, Marquardt H, Reynolds FH Jr, Stephenson JR: Transformation induced by Abelson murine leukemia virus involves production of a polypeptide growth factor. Science 1982;216:894-897.
https://doi.org/10.1126/science.6177040 |
| 233 | Daniel JM, Reynolds AB: The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol 1999;19:3614-3623.
https://doi.org/10.1128/MCB.19.5.3614 |
| 234 | Bocian A, Kędzierawski P, Kopczyński J, Wabik O, Wawruszak A, Kiełbus M, Miziak P, Stepulak A: Kaiso Protein Expression Correlates with Overall Survival in TNBC Patients. J Clin Med 2023;12:370.
https://doi.org/10.3390/jcm12010370 |
| 235 | Vermeulen JF, van de Ven RA, Ercan C, van der Groep P, van der Wall E, Bult P, Christgen M, Lehmann U, Daniel J, van Diest PJ, Derksen PW: Nuclear Kaiso expression is associated with high grade and triple-negative invasive breast cancer. PLoS One 2012;7:e37864.
https://doi.org/10.1371/journal.pone.0037864 |
| 236 | Jones J, Wang H, Zhou J, Hardy S, Turner T, Austin D, He Q, Wells A, Grizzle WE, Yates C: Nuclear Kaiso indicates aggressive prostate cancers and promotes migration and invasiveness of prostate cancer cells. Am J Pathol 2012;181:1836-1846.
https://doi.org/10.1016/j.ajpath.2012.08.008 |
| 237 | Jones J, Wang H, Karanam B, Theodore S, Dean-Colomb W, Welch DR, Grizzle W, Yates C: Nuclear localization of Kaiso promotes the poorly differentiated phenotype and EMT in infiltrating ductal carcinomas. Clin Exp Metastasis 2014;31:497-510
https://doi.org/10.1007/s10585-014-9644-7 |
| 238 | Tian W, Yuan H, Qin S, Liu W, Zhang B, Gu L, Zhou J, Deng D: Kaiso phosphorylation at threonine 606 leads to its accumulation in the cytoplasm, reducing its transcriptional repression of the tumour suppressor CDH1. Mol Oncol 2022;16:3192-3209
https://doi.org/10.1002/1878-0261.13292 |
| 239 | Dai SD, Wang Y, Miao Y, Zhao Y, Zhang Y, Jiang GY, Zhang PX, Yang ZQ, Wang EH: Cytoplasmic Kaiso is associated with poor prognosis in non-small cell lung cancer. BMC Cancer 2009;9:178.
https://doi.org/10.1186/1471-2407-9-178 |
| 240 | Abisoye-Ogunniyan A, Lin H, Ghebremedhin A, Salam AB, Karanam B, Theodore S, Jones-Trich J, Davis M, Grizzle W, Wang H, Yates C: Transcriptional repressor Kaiso promotes epithelial to mesenchymal transition and metastasis in prostate cancer through direct regulation of miR-200c. Cancer Lett 2018;431:1-10.
https://doi.org/10.1016/j.canlet.2018.04.044 |
| 241 | Feng J: Upregulation of MicroRNA-4262 Targets Kaiso (ZBTB33) to Inhibit the Proliferation and EMT of Cervical Cancer Cells. Oncol Res 2018;26:1215-1225.
https://doi.org/10.3727/096504017X15021536183526 |
| 242 | Zhang Y, Jiao H, Wu Y, Sun X: P120-catenin regulates pulmonary fibrosis and TGF-β induced lung fibroblast differentiation. Life Sci 2019;230:35-44.
https://doi.org/10.1016/j.lfs.2019.05.052 |
| 243 | Chen HC, Zhu YT, Chen SY, Tseng SC: Selective activation of p120ctn-Kaiso signaling to unlock contact inhibition of ARPE-19 cells without epithelial-mesenchymal transition. PLoS One 2012;7:e36864.
https://doi.org/10.1371/journal.pone.0036864 |
| 244 | Kahounová Z, Kurfürstová D, Bouchal J, Kharaishvili G, Navrátil J, Remšík J, Šimečková Š, Študent V, Kozubík A, Souček K: The fibroblast surface markers FAP, anti-fibroblast, and FSP are expressed by cells of epithelial origin and may be altered during epithelial-to-mesenchymal transition. Cytometry A 2018;93:941-951 .
https://doi.org/10.1002/cyto.a.23101 |
| 245 | Mehdi AM, Zhou C, Turrell G, Walpole E, Porceddu S, Frazer IH, Chandra J: HPV status represents dominant trait driving delineation of survival-associated gene co-expression networks in head and neck cancer. Cancer Gene Ther 2022;30:629-640.
https://doi.org/10.1038/s41417-022-00577-9 |
| 246 | Ward C, Forrest IA, Murphy DM, Johnson GE, Robertson H, Cawston TE, Fisher AJ, Dark JH, Lordan JL, Kirby JA, Corris PA: Phenotype of airway epithelial cells suggests epithelial to mesenchymal cell transition in clinically stable lung transplant recipients. Thorax2005;60:865-871
https://doi.org/10.1136/thx.2005.043026 |
| 247 | Mižíková I, Palumbo F, Tábi T, Herold S, Vadász I, Mayer K, Seeger W, Morty RE: Perturbations to lysyl oxidase expression broadly influence the transcriptome of lung fibroblasts. Physiol Genomics 2017;49:416-429.
https://doi.org/10.1152/physiolgenomics.00026.2017 |
| 248 | Kirschmann DA, Seftor EA, Nieva DR, Mariano EA, Hendrix MJ; Differentially expressed genes associated with the metastatic phenotype in breast cancer. Breast Cancer Res Treat 1999;55:127-136.
https://doi.org/10.1023/A:1006188129423 |
| 249 | Kirschmann DA, Seftor EA, Fong SF, Nieva DR, Sullivan CM, Edwards EM, Sommer P, Csiszar K, Hendrix MJ: A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res 2002;62:4478-4483.
|
| 250 | Manov I, Hirsh M, Iancu TC, Malik A, Sotnichenko N, Band M, Avivi A, Shams I: Pronounced cancer resistance in a subterranean rodent, the blind mole-rat, Spalax: in vivo and in vitro evidence. BMC Biol 2013;11:91.
https://doi.org/10.1186/1741-7007-11-91 |
| 251 | Odeh A, Dronina M, Domankevich V, Shams I, Manov I: Downregulation of the inflammatory network in senescent fibroblasts and aging tissues of the long-lived and cancer-resistant subterranean wild rodent. Spalax, Aging Cell 2020;19:e13045.
https://doi.org/10.1111/acel.13045 |
| 252 | Friedrich Schiller: Xenien und Votivtafeln. Hofenberg, 1796 ISBN 978-3-8430-7081-2.
|
| 253 | Hammersen F: Histologie, Farbatlas der mikroskopischen Anatomie, 1 Auflage. Johannes Sobotta Verlag, München-Wien-Baltimore, Urban & Schwarzenberg, 1975, ISBN 10: 3541073713.
|
| 254 | Reichert KB: Bericht über die Fortschritte der mikroskopischen Anatomie im jahre 1847, In: Archiv für Anatomie, Physiologie und Wissenschaftliche Medicin, in Verbindung mit mehreren Gelehrten. Dr. Johannes Müller (Herausgeber), Von Veit et Comp. Verlag, Berlin, 1848.
|
| 255 | Lebert H: Physiologie pathologique. Bailiiere, Paris, 1845.
|
| 256 | Virchow R: Die Cellularpathologie in Ihrer Begruendung auf Physiologische und Pathologische Gewebelehre. Hirschwald A Verlag, Berlin, 1858.
|
| 257 | Fox W (1864) On the origin and mode of development of the cystic tomonrs of the ovary. Med Chir Transact Vol 157:June 28th 1864.
|
| 258 | Ziegler E: Lehrbuch der Allgemeinen und Speciellen Pathologischen Anatomie und Pathogenese. Verlag von Gustav Fischer, Jena, 1881.
|
| 259 | Russell W (1890) An Address on a Characteristic Organism of Cancer. Br Med J 2:1356-1360.
https://doi.org/10.1136/bmj.2.1563.1356 |
| 260 | Cajal SR: Manual de anatomı'a patolo' gica general: seguida de un resumen de microscopia aplicada a la histologı'a y bacteriologı'a patolo' gicas, Second Edition (N. Moya), 1896.
|
| 261 | Loeb L: Über die Entstehung von Bindegewebe, Leucocyten und roten Blutkörperchen aus Epithel und eine Methode, isolierte Gewebsteile zu züchten. Max Stern & Co., Chicago, 1897.
|
| 262 | Maximow AA: Experimentelle Untersuchungen über die entzündliche Neubildung. G. Fischer Verlag, Jena, 1902.
|