Kca3.1-Related Cellular Signalling Involved in Cancer Proliferation
bCentro de Biociencias, Fundación Instituto de Estudios Avanzados-IDEA, Estado Miranda, Venezuela,
cLaboratorio de Cultivo de Tejidos y Biología de Tumores del IBE, Facultad de Ciencias, Universidad Central de Venezuela
Keywords
Abstract
Introduction
Cancer diseases are characterized by genetic instability, resulting in a series of changes in cell physiology called hallmarks [1, 2], that provoke malignant uncontrolled proliferation [3]. Many of those alterations involve the activity of ion channels [4]. Specifically, potassium channels are involved in the modulation of cell cycle progression, leading to uncontrolled proliferation [5]. Potassium channels are abnormally expressed in several types of cancer tissues and cell lines [6]. They are also involved in controlling the progression of cell cycle, both through permeation-related phenomena and non-canonical permeation-independent mechanisms [5]. Regarding permeation-dependent mechanisms, membrane potential changes seem relevant to determine cell cycle progression. Potassium channels determine membrane potential (Vm) changes through the cell cycle [7, 8]. Non-canonical mechanisms generally involve the interaction of cytoplasmic domains of potassium channels with other proteins to exert some functional roles during the oncological process, independently of their permeating capacity [9, 5]. Potassium channels are relevant in intracellular signaling pathways by any of the two mechanisms mentioned above. K+ channels are also relevant to cancer because of their interaction with many physiological variables. Some cancer-involved K+ channels are responsive to transmembrane voltage changes (Kv) [10], while others respond to some signaling molecules and physiological variables such as oxygen tension, pH, and mechanical stretch as the two-pore-domain channels(K2P) [11, 12]. In this sense, calcium concentration sensible K+ channels (KCa) are involved in cancer signal transduction in more ways than in their interaction with calcium [13], as mentioned below. Remarkably, this kind of channel has been found co-localized with calcium channels and appears to contribute to some of their hallmarks [14]. KCa channels are significant in proliferation-associated signaling because cell cycle progression depends on Ca2+ signal changes [15, 16].
All eukaryotic cells require Ca2+ signaling for cell proliferation, but some transformed cells and tumor cell lines exhibit a reduced Ca2+ dependence. Growing evidence shows that Ca2+ signaling pathways are often remodeled or deregulated in cancerous processes to sustain proliferation [17, 18, 19]. KCa channels are significant pieces in the deregulated proliferation via interaction with signaling molecules, as is the case with the big-conductance calcium-activated channel (BKCa) [20], or KCa3.1, which is the subject of this review.
KCa3.1 is involved in cell cycle signaling of various cell types, either through permeating evoked calcium signals or non-canonical ways. The Ca2+-activated K+ channel KCa3.1, also known as SK4, IK1, and IKCa1, encoded by the KCNN4 gene, has been implicated in physiologically relevant processes related to cancer hallmarks such as abnormal proliferation, metastasis, avoidance of apoptosis, support of angiogenesis and epithelial-to-mesenchymal transition [13]. Besides, this channel is up-regulated and promotes proliferation in some cell lines and their respective cancerous tissues, compared to their adjacent noncancerous surroundings. In particular, KCa3.1 takes part in the control of cell cycle progression in several tissues, such as: human pancreatic cancer cell lines [21], human endometrial cancer [22], human breast cell line MCF-7 [23], breast cancer murine model [24], prostate cancer cells [25], mesenchymal stem cells [26], colorectal [27], and finally liver, were it has also a role in cancer progression [28]. Although this channel is involved in cell cycle control, the complete mechanisms are unknown. This channel is involved in physiological mechanisms of cancerous or normal cells that allow proliferation, such as energy management [29], membrane potential and volume control [30, 31, 32, 33].
This review addresses the elucidation of the intermediary steps between either the expression or activity of this channel and the signals that promote cell proliferation [34, 35, 36, 32, 28, 37]. This channel is also relevant in cancerous progression and as a marker and prognostic tool [38]. The goal of this review is to relate the functioning of this channel within the cell proliferation signals in both non-cancerous and tumoral cells. It addresses the participation of this channel in some mitogen proliferative pathways, including calcium-related signaling and the non-canonical interactions. In particular this review states some details about deregulated proliferating pathways involving KCa3.1 in cancerous cells. The contrasting differences between cancerous and normal signaling involving this channel may aid in the development of anticancer therapies. Before discussing signaling processes, we first address some characteristics of KCa3.1.
Some features of the KCa3.1 and its functional control
KCa3.1 structure and general functional features
KCa3.1 is a homo-tetrameric voltage-independent K+ channel with six
transmembrane segments, which is constitutively associated with calmodulin
(CaM) at the intracellular C-terminal CaM-binding domain, and it has the pore
motif between the 5th and 6th segment. The CaM C-lobe binds in Ca2+-independent
manner to two alpha helices (A and B) at the C-terminal cytoplasmic region,
just beside S6 in the C-terminal direction (CAMBD1, K(312)-T(329) or C-L-CaMBD
in Fig. 1). During channel gating when [Ca2+] rises, the CaM N-lobe
binds to an N-Lobe CaM binding domain in a nearby subunit of the channel,
different from that subunit binding the C-lobe. That N-Lobe CaM binding domain
is one of the two helixes conforming the S4-S5 linker proximal to S4 (S45A
helix: N-L-CaMBD in the Fig. 1). A detail structural description is given by
Lee and MacKInnon [39].
This channel has a selectivity for K+ more than ten times higher than for
Na+, and its cooperative sensitivity to [Ca2+]i
establishes a Hill relationship with the maximal open probability of 0,44 above
1 μM [Ca2+]i, and the following affinity and Hill
coefficient parameters respectively: KD = 188 nM and nH =
3.2 [40]. The single conductance of this
channel ranges between 20 to 80 pS, hence intermediary (IK) among
single-channel conductance of other Ca2+ activated K+
channels (KCa): e.g., small conductance KCa (SK, 5–20 pS) and big-conductance
KCa (BK, 100–300 pS) [41]. In addition to Ca2+, other signaling
molecules modulate channel gating, which will be discussed in the following
section.
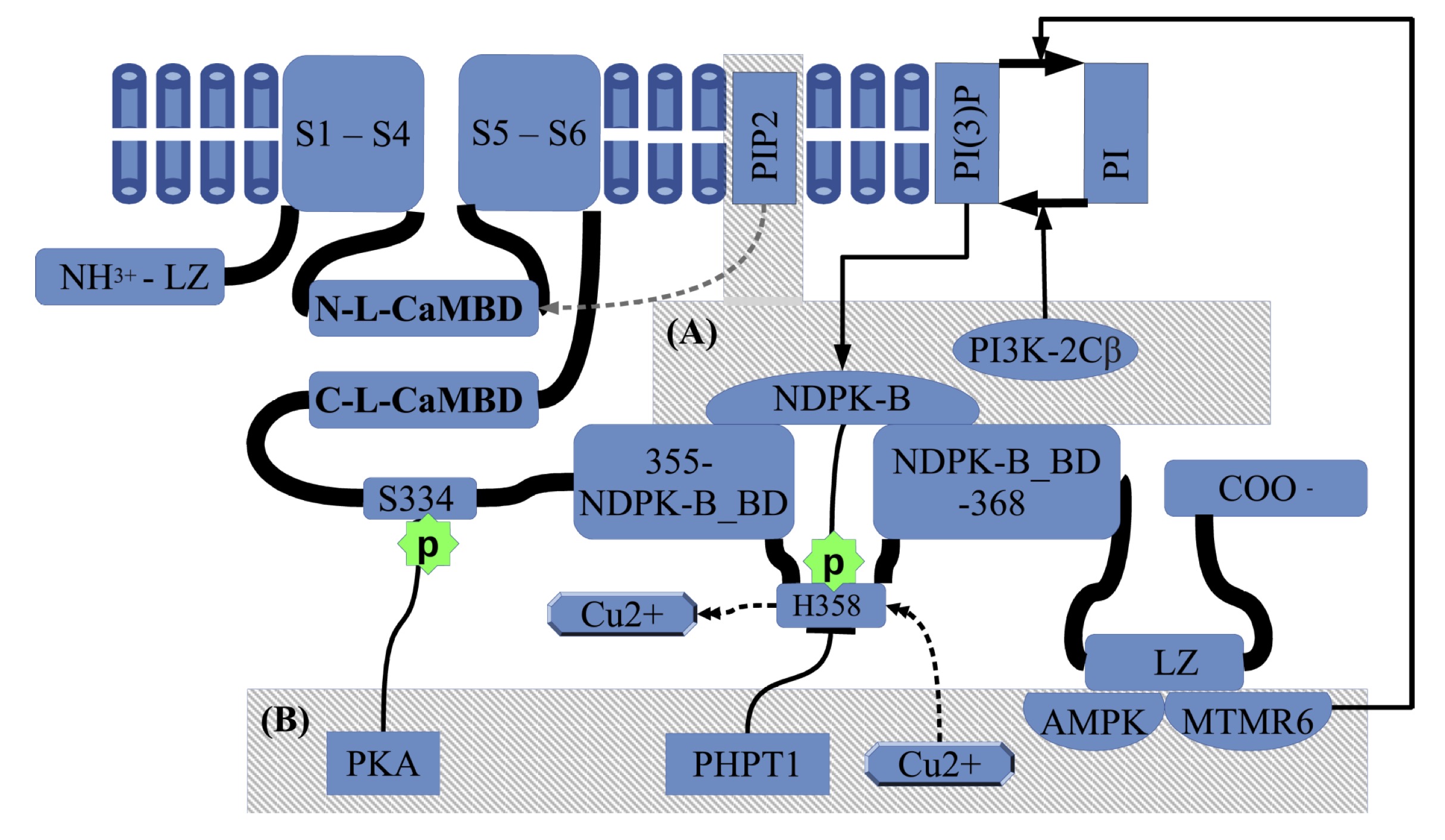
Fig. 1: Regulation of KCa3.1 activity by natural signaling molecules. The general structure of KCa3.1 consist of an intracellular N-terminal domain, a transmembrane segment that contains six transmembrane helices(S1 to S6). The selectivity filter is included between the 5th and the 6th transmembrane helices(THs). Between S4 and S5 there are two linker helices, the closer to S4 is called S45A, which is the N-Lobe CaM binding domain (N-L-CaMBD). Coming from the THs containing segment the C-terminal has one C-Lobe-CaM binding domain (C-L-CaMBD), followed by an intermediate inhibitory phosphorylation site on S334 residue. On the way to C-terminal there is a NDPK-B binding domain(NDPK-B BD), which includes the H358 phosphorylating site, involved in activation. The copper inhibition is mutually exclusive with H358-phosphorylation. Also, the PI(3)P-PI equilibrium modulates the NDPK-B activating action. Finally, closer to the C-terminal there is a LZ domain working as an anchor site for AMPK and MTMR6. The (A) box summarizes the activating kinases and the recently reported of PIP2 activator which acts at the N-Lobe-CaMBD, and the (B) box corresponds to inhibitory factors.
KCa3.1 activity regulation
The positive modulators tend to increment the open
state channel probability (PO), while negative ones do the opposite. Some
molecules are known to directly regulate the channel activity through
phosphorylation of specific sites [42, 43, 38]. Others may include interaction
through intermolecular forces [44, 45] or upstream regulation of the direct
interacting molecule [46].
KCa3.1 positive modulators
In addition to Ca2+,
phosphorylation of the His358 residue (h358) can activate KCa3.1. The
nucleoside diphosphate kinase B NDPK-B phosphorylates h358 and releases a
coordinated Cu2+ that causes channel inhibition [43] (see Fig. 1).
NDPK-B binds to KCa3.1 through C-terminal domain (R (355)-M (368)) (Fig.1) and
seems to act as a downstream target of phosphatidylinositol-(3)-phosphate
(PI(3)-P), generated by the PI3 kinase subclass type IIβ (PI3K-2Cβ) [47, 48, 49].
Finally, recently has been reported the phosphatidylinositol 4, 5-bisphosphate
(PIP2) as a potent KCa3.1 activator that acts at the interface of the
CaM-binding domain [50] (see Fig. 1). ATP also
activates KCa3.1 through its C-terminal domain, with residues from Arg(355) to
Ala(413) being necessary and sufficient. Direct ATP binding is unlikely as the
involved mentioned region lacks consensus ATP binding sites [49]
KCa3.1 negative modulators
Negative regulation of the channel can be achieved by dephosphorylation of the p-h358 residue, which is operated by the phosphohistidine phosphatase-1 (PHPT1: see Fig. 1) [51]dephosphorylation of PI(3)-P by myotubularin-related protein 6
(MTMR6) [52]. In addition, intracellular AMP activates a 5’AMP-activated protein kinase
(AMPK) that seems to regulate KCa3.1 through protein-protein interactions [44].
MTMR6 and AMPK have protein-protein interaction in the
C-terminal Leucine
Zipper motifs (LZ, Fig 1) Finally, the
cAMP-activated protein kinase A(PKA) can phosphorylate Ser334(s334), which
decreases the open probability of KCa3.1 by reducing CaM binding [42].
KCa3.1 inhibitors and activators
Inhibitor compounds have been tested in model systems and clinical trials for several
pathological conditions. They are powerful tools to interact with malignant
cell cycle progression and thus tumor growth, cell migration, and apoptosis
scape, among other hallmark-related phenomena [13]. Charybdotoxin (ChTx) and
maurotoxin (MTx) are high-affinity peptide toxins against KCa3.1. However, MTx
has the highest affinity for KCa3.1 while being highly specific below 1μM [53].
Among the small synthetic inhibitors, TRAM-34 is an agent with improved
properties (1-[(2-chlorophenyl) diphenylmethyl]-1H-pyrazole), which is a
modified triarylmethane pyrazole analog of the clotrimazole molecule with higher
affinity [54] although, it inhibits some human isoforms of cytochrome P450
(CYP) [55]. Finally, senicapoc is the highest affinity (IC50: 11 nM) known
inhibitor [56], as shown in Table 1. Regarding KCa3.1 activators, there are two
non-selective ones: 1-EBIO (1-ethyl-2-benzimidazolinone) [57], DC-EBIO (5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one)
[58], the highly potent NS-309(EC50: 20 nM) (3-Oxime-6,7-dichloro-1H-indole-2, 3-dione)
with very short in vivo half-life and low selectivity [59]; SKA-31 which
is more selective [60], with relatively high affinity (EC50: 250 nM) [61]; and
finally the most selective and potent to date is SKA-121(EC50: 111 nM) [62], as
shown in Table 2. Inhibitors and activators are available to interact with
KCa3.1, making it possible to dissect its functionality in cancer cells.
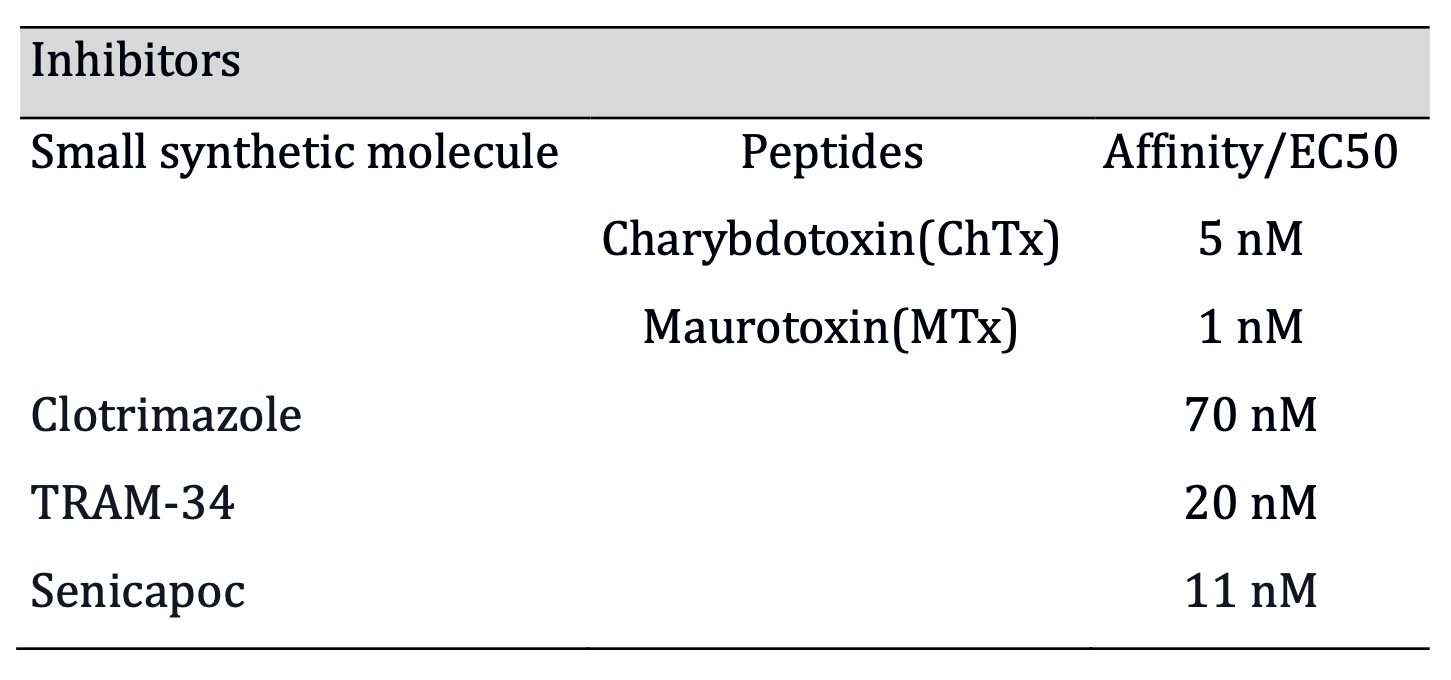
Table 1: Senicapoc has the highest affinity among the synthetic small molecule inhibitors, and ChTx has hightest affinity of all (Affinities for Mtx, ChTx, TRAM-34, clotrimazole, extracted from Wei et al., 2005; Senicapoc from Stocker et al., 2003)
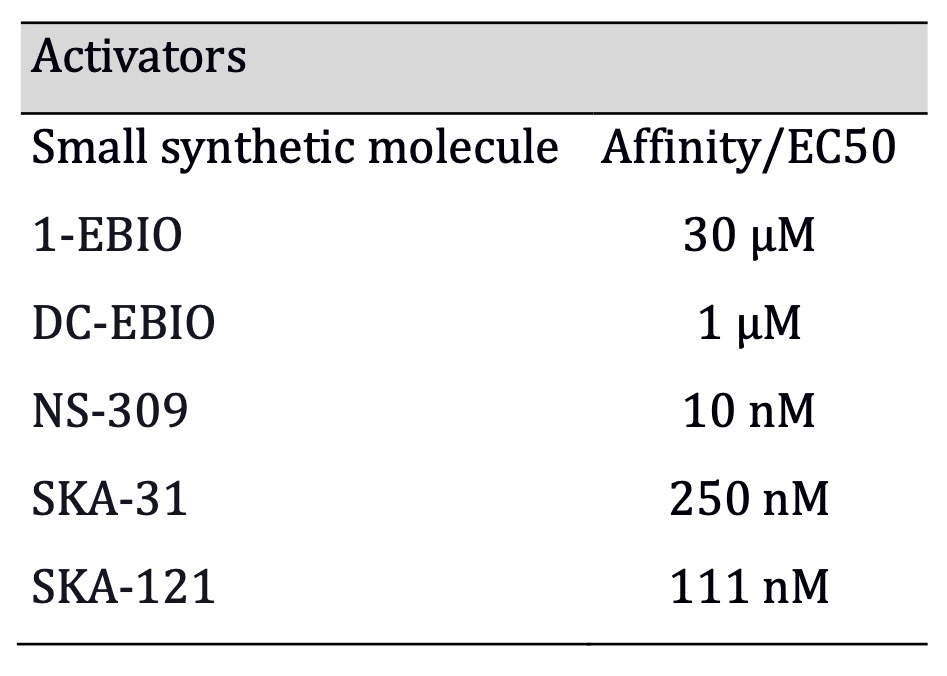
Table 2: SKA-111 combines hight affinity with hight specificity among all activators (Affinities for activators were obtained: 1-EBIO from Wulff, and Köhler 2013; DC-EBIO from Wei et al., 2005; SKA-31 from Sankaranarayanan et al., 2009; NS-309 from Strøbaek et al., 2004; SKA-121 from Coleman et al., 2014)
KCa3.1 trafficking
The assembly and trafficking of KCa3.1 to the cell surface
require CaM binding in addition to gating [63]. This process is inhibited by
cAMP kinase (PKA) acting on a single phosphorylation site [46]. The LZ consensus regions, which are composed of
repeating heptads containing leucine at the C(L399/L406) and N(L18/L25) termini
are required for proper anterograde trafficking to the plasma membrane [64,
65], in addition to the S4-S5 linker, and Lys197 (K197) necessary for the
release from ER [66]. According to Schwab et al [67]., retrograde trafficking,
which involves endocytosis from the plasma membrane, is facilitated by clathrin
and a C-terminal dileucine motif. At the plasma membrane, KCa3.1 is mildly
ubiquitylated; after endocytosis, it is polyubiquitylated. Finally, the rate of
lysosomal degradation determines deubiquitylation through UPS8
(ubiquitin-specific protease) [68]. The process is accomplished through the
Rab7 small-molecular-weight guanine nucleotide-binding protein and the
endosomal sorting complexes required for the transport (ESCRT) pathway [69].
Before trafficking to the plasma membrane, the expression of this channel is
controlled by molecular mechanisms discussed in the following chapter.
Control of KCa3.1 expression in cancerous cells
Basic control of KCa3.1 expression
The transcription of KCa3.1 is regulated by at least two factors: Activator Protein
(AP-1) and Repressor Element 1(RE1)-Silencing Transcription factor (REST) [70].
In T-lymphocytes the continuing cell proliferation and the transcription of
KCa3.1 require Activator Protein (AP-1) and Ikaros-2 activity, triggered by the
PKC mitogenic pathway [71]. REST binds to the RE-1 site in the KCNN4 gene in
vascular smooth muscle cells (VSMCs), repressing its transcription. The
decrease in REST expression allows KCa3.1 expression and facilitates
proliferation. That is consistent with the detected KCa3.1 activity in
proliferating VSMCs but not in quiescent VSMCs. AP-1 is a potential
transcriptional activator that may be coordinated with REST to regulate KCNN4
expression in VSMC [72] (See Fig. 2: F).
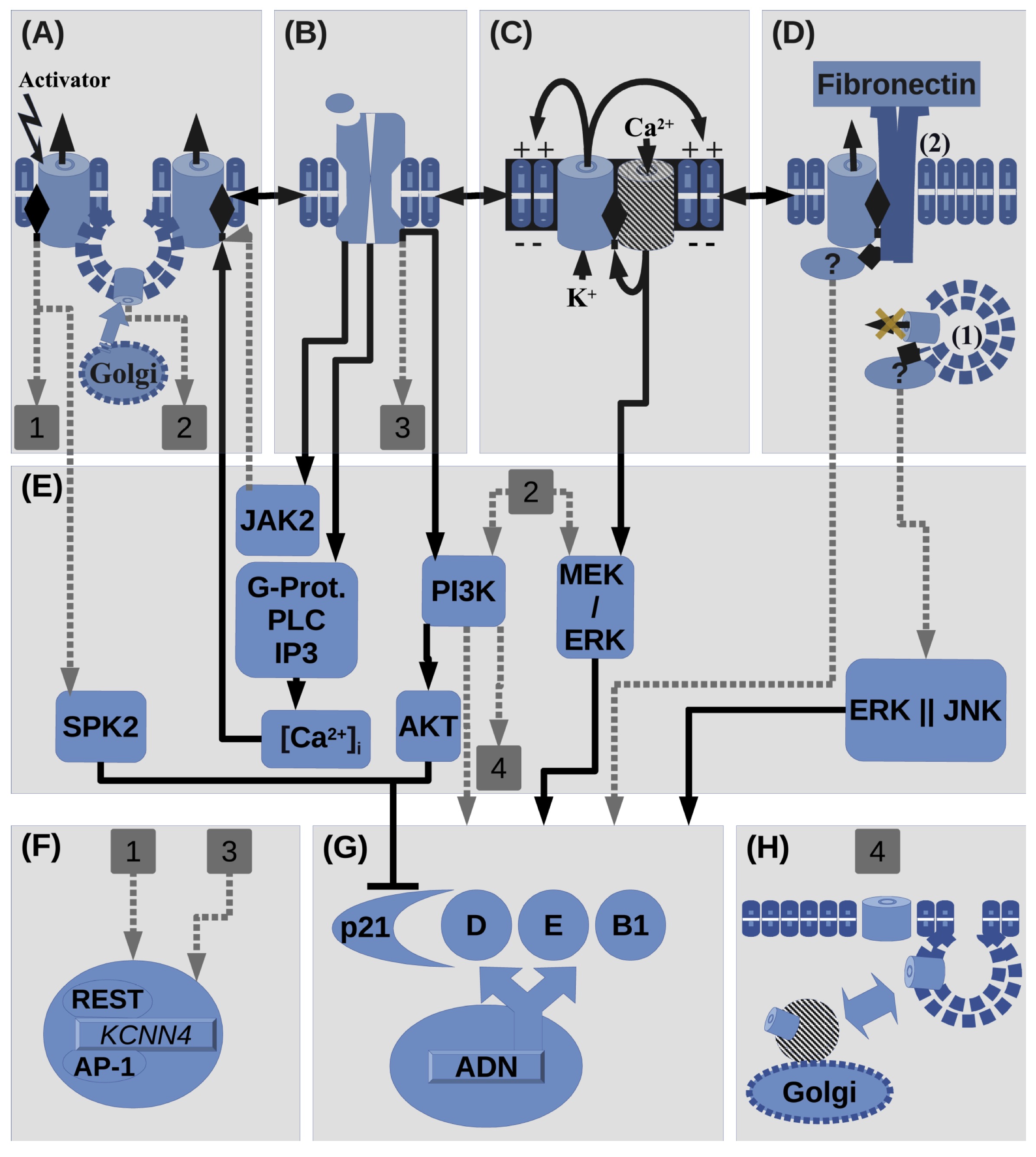
Fig. 2: Summary of interactions involving KCa3.1 in proliferation. (A) Activation(e.g. 1-EBIO) of KCa3.1 and stimulation of its surface expression, by e.g. interaction with plasma membrane(PM) receptors (PMR, shown in B). KCa3.1 activation site is represented in the channel cartoon as a lateral diamond symbol. KCa3.1 can be activated by Ca2+, JAK2 and its regulating kinases(shown in Fig. 1). KCa3.1 activation can promote its transcription(Shown by square label 1) or SKP2 activation, which promotes cell cycle by inhibiting p21. Also KCa3.1 surface up-regulation/expression, which can be regulated by receptors in B, can promote proliferation through PI3K/Akt and MEK/ERK pathways(Shown by square label 2: which corresponds to label 1 in F). (B) Histamine/Hormone/Growth factor Receptors(PRL, GFRs: IGF-1, EGF, TGF-β, HGF) can regulate activity of A or C(cooperating channels). Vice versa relation among A, B and C, is also possible. Mitogens can up-regulate KCa3.1 transcription(in the case of TGF-β, shown by square label 3) or activate KCa3.1(in the case of PRL) through JAK2, which also promotes proliferation(omitted pathway because of its indetermination and graphical overload). Also PMR can regulate KCa3.1 trafficking through IP3K pathway (shown by square label 4). (C) Positive feedback between KCa3.1 and Ca2+ channels(CaC). KCa3.1 drives membrane potential(Vm) while CaC drives increments in [Ca2+]i, including SOCE. CaC can be: TRPC, TRPV, Orai, or CD20. This phenomenon may interact with B(interaction with PMR) and D(non-canonical functions). Ca2+ entry can regulate ERK phosphorylation.(D) Non-canonical functions: in case (1), KCa3.1 expression seems to promote proliferation while in traffic to the PM, through ERK and JNK pathways in parallel. In the case (2), interaction between KCa3.1 and β-1 integrins in presence of fibronectin promotes proliferation. As TRPC1 expression is also stimulated by interaction with fibronectin the cooperation with KCa3.1 is possible. KCa3.1,TRPC1 and integrin expression is also promoted in presence of fibronectin. (E) Summary of intracellular signaling molecules that involve KCa3.1 in proliferation. (F) KCa3.1 gene expression can be regulated by undetermined pathways involving PMR and KCa3.1 activation at the membrane. (G) Expression and activity of cyclins or its CDKIs. As far as can be pointed on this review KCa3.1 can stimulate cell cycle advancement by MEK/ERK, and by SPK and PI3K/AKT pathways through p21 inhibition. (H) KCa3.1 expression and anterograde/retrograde traffic can be regulated through signaling pathways, in particular PI3K.
Deregulation of KCa3.1 expression
Abnormal expression of K+ channels can occur in
tumors at various levels, including genomic, transcriptional,
post-translational, or epigenetic [73]. In
gliomas, e.g., the KCa3.1 potassium channel intervene in several malignancy
aspects, including proliferation [74]. In glioblastoma cells, GL-15, the
expression of KCa3.1 is regulated by the ERK1/2 pathway, which is often
deregulated in cancerous cells [40]. Consistent with this, as found by Park et
al [75]., in mouse aortic endothelial cells (MAECs), the KCa3.1 expression is
promoted by AP-1 via ERK (see Fig 2 A, Label 1) and repressed by REST. KCa3.1
is typically overexpressed in many cancerous cell lines, including human breast
cancer cells MDA-MB-231 [76] and MCF-7. Its expression varies as the cell cycle
advances, as evidenced by the latter [23]. Even though mechanisms that give
rise to altered expression of ion channels are not well understood, it is
believed that maintained expression of specific potassium channels in cancerous
cells may confer a selective advantage [77, 6].
The epigenetic control of KCNN4 expression is
performed by modification of the pattern in chromatin acetylation or
methylation and DNA methylation close to the gene. In the advanced-stage tumors
of non-small cell lung cancer, KCa3.1 mRNA over-expression is due to gene
promoter hypomethylation, compared to normal tissue [78]. A posterior study in
cervical tissue suggests that over-expression results from positive feedback in
which influx, promoted by KCa3.1-operated
hyperpolarization, activates AP-1 and post-translational histone acetylation.
Thus, further expression of the channel. Histone acetylation within the
promoter enhances transcription factor binding, thereby maintaining an open DNA
conformation for the transcription factor to access [79]. Furthermore, histone
deacetylase inhibitors (HDACis) down-regulates KCa3.1 transcription in breast
(YMB-1) and prostate (PC3) cancer cell lines [80]. In addition,
other up-regulation mechanisms have been uncovered: e.g., in neointimal
hyperplasia produced by proliferating vascular cells, the KCa3.1 de
novo transcription is promoted through the reduced expression of
REST acting on RE1 sequence in KCNN4 [82]. REST plays different roles in normal and
cancerous proliferation depending on cell type considered. That is to say, in
the nervous system and its tumors, REST acts as an oncogene, whereas it acts as
a tumor suppressor in some carcinomas of breast, lung, and colon [83, 84].
Another feature is that in HaCaT keratinocytes and C6 glioma cells, the
expression of the human KCa3.1 channel seems to be regulated by a feedback
mechanism since after three days of treatment with 1-EBIO, mRNA levels and
activity of human KCa3.1 were substantially diminished, accompanied by loss of
mitogenic activity and an increase in cell size, all in a reversible way [85].
Intracellular signals may determine the expression of this channel and its
feedback mechanisms of expression. Understanding those processes may lead
someday to the regulation of cancer proliferation. In particular, studying the interaction of this channel
within the signaling network may improve the understanding of its proliferative
role in both cancer and normal cells.
Control of proliferation by extracellular signals through expression and/or activity of KCa3.1
Depending on the cell line, KCa3.1 expression has different influences upon
proliferation. For instance, in vitro assays
show that over-expression or activation of this channel increases proliferation
in human prostate cancer cell lines [25], whereas it seems innocuous for
proliferation in human breast cancer cell line MDA-MB-231. Moreover, KCa3.1
activation decreased proliferation in non-tumorigenic immortalized breast cell
line MCF-10A, even in control or KCa3.1 over-expressing cells, but not by
KCa3.1 over-expression alone. Surprisingly, KCa3.1 over-expression increased
the primary tumor growth of MDA-MB-231 in orthotopic xenografts. In breast
cancer cells, evidence suggests that microenvironmental factors influence
KCa3.1 activation and its signaling pathways, which may be significant for
extracellular signal-dependent proliferation [76]. Different proliferative
influences of KCa3.1 should correspond to the particular signaling network of
each cell line and the environmental differences, which might be related to
extracellular factors: e.g., in breast cancer murine models, this channel is
involved in mitogenic-dependent cell cycle promotion, since serum-containing
growth factors evoked Ca2+ signals and
the transition to S phase was suppressed by pharmacological blockade or genetic
ablation of KCa3.1 [24]. Old data also suggest that KCa channels are involved
in the response of breast cancer cells to extracellular factors such as
EGF(Fig.2, A-B) and insulin [86, 87].
KCa3.1 channel activity can increase by
upstream changes, such as the activation by hormones/growth factor receptors
(Fig. 2, A-B) [74]. This channel plays roles at
different levels within the mitogen-activated signaling chain. In breast cancer cells, Ca2+ -activated K+ channels may play their role in
cell cycle physiology through the mitogen-initiated electrical signals. EGF
treatment of mouse mammary cells induced transient hyperpolarizations of the
membrane potential mediated by KCa channels [86]. Since the last decade of the
20th century, evidence shows that in the estrogen receptor-positive (ER+)
breast cancer cell line MCF-7, a channel with all the characteristics of KCa3.1
is upregulated during its proliferative phase compared to cells in the plateau
phase or tamoxifen-treated cells. However, not all cells
rely on this channel to proliferate, as it is absent in some regularly
proliferative cell batches [88]. There are alternative pathways for
proliferation, and the KCa3.1 channel may play a role in at least one of these
signaling pathways mediating proliferation.
Besides, in the same cell line insulin-like
growth factor 1 (IGF-1) mitogenic effect, mediated by Akt signaling pathway,
depends on the Kv10.1 voltage-dependent K+ channel [89]. More
recently, Kv10.1 and KCa3.1 have been involved in MCF-7 basal proliferation but
surprisingly not in estrogen-stimulated proliferation [90]. As MCF-7 cells
express both estrogen receptors, ER-alpha and ER-beta [91], proliferation is much
faster when treated with estrogens [92], but at least this mitogen’s associated
signaling does not seem to involve those channels. Interestingly, in this cell
line (MCF-7), TRAM-34, a supposedly specific KCa3.1 blocker, has been reported
as a novel non-steroidal ER agonist [90]. It remains to see whether there are
any K+ channels involved in MCF-7 estrogen-triggered
proliferation. Regarding the proliferative role, KCa3.1 mRNA
expression in tumor cells is significantly correlated, on one side by the
absence of estrogen receptor (ER-)/progesterone receptor (PR) status and the
P53 abnormal gene on the other side Those features
are unfavorable breast cancer parameters [93]. On the other side, another
well-known mitogen in breast cancer cells is prolactin (PRL), which induces
proliferation by increasing KCa3.1 activity through the Janus Kinase 2(JAK2)
signaling pathway (See Fig 2. A-E-B). Human KCa3.1 channels constitute a target
of PRL, leading to breast cancer cell proliferation [34]. The JAK2/STAT5
pathway is involved in MCF-7 cell proliferation by activating the cyclin D1
gene promoter [94]. Multiple studies indicate that PRL, which could be within
circulating plasma or expressed locally by mammary tissue, among others,
promotes proliferation and metastasis in various breast cancer models, in
vitro and animal models [95]. Specifically in non-PRL expressing
MCF-7 derived cells, the PRL receptor (PRL-R) stimulates the cell cycle by
increasing cyclin D1 and B1 expression and facilitates the G1/S transition by
decreasing the p21(WAF1) levels [96]. The relationship of KCa3.1 to these cell
cycle regulators is unknown in breast cancer cells but has been reported in
other cell types, as discussed in Section 5.
Growth factor-induced proliferation also
appears to be associated with KCa3.1 expression. For instance, in respiratory
smooth muscle, KCa3.1 channel upregulation is mitogen stimulated: in human
airway smooth muscle (HASM) cells by transforming growth factor-beta (TGF-β)
and in human bronchial smooth muscle (BSM) by platelet-derived growth factor
(PDGF), as evidenced by increases in mRNA, protein expression and activity [97,
98]. Furthermore, the expression of KCa3.1 also switches BSM cells from a
contractile to a proliferative phenotype through activation of ERK and PI3K/Akt
pathways (Fig. 2, A-E, Label 2) [98]. In lung carcinoma, KCa3.1
expression promotes lung adenocarcinoma cell proliferation through the PI3K/Akt
and MEK/ERK signaling pathways (Fig. 2, A-E, Label 2) [37]. These pathways are
also active in non-small cell lung carcinoma cell lines (NSCLC) and determine
EGF-dependent proliferation [99].
In Daudi lymphoma B cells, the intermediate
conductance KCa channel was upregulated on the cell surface during cell cycle
progression in response to fetal bovine serum (FBS) via a phosphatidylinositol
3-kinase (PI3K)-dependent pathway, possibly through the IGF-1 receptor, as it
is involved in PI3K-associated pathways. The trafficking of this channel can be
mediated by PI3K, as has been shown for other channels (Fig. 2, B-E, Label 4)
[100]. PI3K controls intracellular vesicular traffic, specifically class II and
class III regulate membrane traffic in endosomal recycling (Fig. 2, pathway
E-H, Label 4) [101]. Besides, in human gastric adenocarcinoma cell line SC-M1
cells, Ca2+ activated and voltage-independent K+ current,
and therefore possibly carried by KCa3.1, is involved in hepatocyte growth
factor (HGF)-induced cell proliferation, even though it did not correlate with
the density of HGF receptors [102]. Finally, another external factor is
histamine, which has a crucial role in cancer progression, stimulating cancer
hallmarks in cell lines derived from breast, pancreas, and hepatoma [103],
particularly modulating the proliferation and migration of glial cell lines
derived from brain tumors [104, 105]. Histamine selectively activates the
KCa3.1 channel in human glioblastoma GL-15 cells by histamine receptor(H1)
activation and G protein/PLC/IP3/[Ca]i pathway (Fig. 2, B-E-A). The high Ca2+ affinity of KCa3.1 confers its selectivity, although there
are other Ca2+ -sensitive channels [106]. As mentioned above, the evidence points to
KCa3.1 as a participant in the proliferative signaling pathways that depend on
diffusible mitogenic factors. The following section discusses the
proliferative processes that regulate the cell cycle through intracellular
signaling involving KCa3.1.
KCa3.1 regulates some kinase proteins within cell cycle signaling
The cell cycle course is controlled by cyclin proteins through its Cyclin-Dependent Kinases (CDKs) [107] and also by Cyclin-Dependent Kinase Inhibitors (CDKIs) [108]. In particular, cyclins D1 and E allow getting through the G1 and G1/S transition. In human normal BSM and murine mesenchymal stem cells (MSCs) , KCa3.1 activation promotes c yclin D1 expression [109; 98]. Moreover , blocking KCa3.1 expression or activity in cancerous cells, such as angiosarcoma cell line ISO-HAS and human endometrial carcinoma (EC) cells HEC-1-A and Ishikawa, reduce expression levels of cyclin D1 and in the two last ones also cyclin E decreases [22, 110].
In human prostate cancer cells (PCa), KCa3.1 channel inhibition leads to an accumulation of p21Cip1 CDKI [111], a cell cycle regulator involved in cell cycle arrest in response to stressful stimuli, transcriptional regulation, modulation, or inhibition of apoptosis and differentiation [112, 113]. A recent report on hepatocellular carcinoma (HCC) cells suggests that KCa3.1 promotes cell cycle progression through post-transcriptional regulation of S-phase protein kinase 2 (SKP2) expression, which subsequently promotes p21/p27 ubiquitin-mediated degradation ( Fig. 2, pathway A-E-G) [28]. KCa3.1 may participate in cell-cycle control by positively regulating G1 cyclins and negatively regulating CDKIs. Besides, the PRL pathway in breast cells interacts with D1 cyclin and KCa3.1, as seen above. It remains to know what is the causal sequence and whether it is the same as in other cell lines, such as HCC. Finally, in glioblastoma tumor cells, KCa3.1 regulates the passage through the G2/M checkpoint via the cdc25C (cell division cycle 25C) phosphatase activity [32]. This phosphatase triggers entry into the M(mitosis) cell cycle phase by dephosphorylating the cyclin B-Cdk1 [114]. The relationship between the KCa3.1 channel and the phosphorylation dynamics that control the cell cycle is beginning to be elucidated. Similarly, the electrophysiological effects on the cell cycle are also evident, as shown below.
Nonspecific effects of KCa3.1 inhibitors upon proliferative capacity
Some cancer cells express the KCa3.1 channel, which is not involved in proliferation, although KCa3.1 inhibitors affect proliferation. In glioblastoma cell lines (U251 and U67) and primary glioma cultures, where KCa3.1 was found functionally expressed, it is not involved in proliferation, as demonstrated by knocking down this channel with siRNA. Nevertheless, using KCa3.1 inhibitors (clotrimazole and TRAM-34), an antiproliferative effect was obtained, but only at concentrations higher than those necessary to inhibit the channel. Therefore, the authors concluded that the effect on proliferation is due to off-target actions [115]. Coincidentally, in human IGR1 melanoma cells expressing KCa3.1, the proliferation was not affected by channel inhibition [116]. Other studies on melanoma and pancreatic cancer cell lines also suggest that inhibitors of this channel (clotrimazole or senicapoc) may act on targets outside the plasma membrane or perhaps block IKCa3.1 channels in intracellular organelles [117]. As a matter of fact, TRAM-34 has been reported as an agonist of ER nuclear receptor, in the breast cancer cell line MCF-7 [ 90 ].
In addition to the plasma membrane, functional expression of mitochondrial KCa3.1(mtKCa3.1) also has been identified on the inner mitochondrial membrane of several cancer cell lines: non-small cell lung cancer (NSCLC) cells [118], pancreatic ductal adenocarcinoma (PDAC) cells [119], colon cancer cells [120], melanoma cells [121] and cervical cancer cells. Although there is some evidence that mtKCa3.1 may be involved in proliferation, it is inconclusive [122] and cannot exclude the off-target effect [117]. Targeting this channel with novel mitochondria-targeting TRAM-34 derivatives reduced tumor growth and metastasis in orthotopic melanoma and pancreatic ductal adenocarcinoma models [123]. In pancreatic ductal adenocarcinoma cell lines, there is evidence that mtKCa3.1 is involved in mitochondrial respiration and proliferation. However, the authors do not exclude that the effect on proliferation could be due to plasma membrane-expressing channels or off-target effects of the KCa3.1 channel inhibitors. This mitochondrial expression of the channel, in contrast to normal cells, may be another reason to address the therapy using this target. Nevertheless, it is unclear whether the proliferation of pancreatic cancer cells depends on mtKCa3.1 [119]. Instead, the mtKCa3.1 channel seems to play a role in the response of melanoma cells to apoptotic stimuli [124]. In melanoma cells (WM266-4) and pancreatic cells (Panc-1), clotrimazole and senicapoc decreased viability equally, even though the former expresses the KCa3.1 channel depending on the cell cycle phase and the latter poorly expresses it. The reduction in viability by the KCa3.1 inhibitor was not a direct result of its blockade at the plasma membrane. Therefore, effects through the channel expression/activity on intracellular membranes cannot be excluded. The use of the KCa3.1 inhibitor (BA6b9) did not affect the viability of Panc-1 cells, even at high concentrations. However, it had slight viability effects on WM266-4 cells, indicating potential secondary targets for the former KCa3.1 inhibitors or effects that depend on the expression of the channel at intracellular organelles. It is important to note that this mechanism strongly depends on the chemical nature of the drug [117]. The evidence linking the mtKCa3.1 channel to proliferation is limited and inconclusive.
KCa3.1 inhibitors affect proliferative capacity on irradiated glioma cells
Plasmalemma KCa3.1 regulates the cell cycle in
irradiated tumor cells. In human glioblastoma (GB) cell lines (T98G, U87MG),
KCa3.1 is upregulated in the plasma membrane and also in glioma during
transformation and malignant progression, in contrast to its low expression or
absence in astrocytes, a type of glia in the central nervous system. In these
GB cell lines, ionizing radiation activates KCa3.1, which increases [Ca+2]i
and induces G2/M arrest. Presumably, the increase in [Ca+2]i
is involved in this activation. Furthermore, pharmacological inhibition of
KCa3.1 (clotrimazole or TRAM-34) or its mRNA knockdown in irradiated cells
reduced IR-induced G2/M arrest, DNA repair, and clonogenic survival. In
addition, inhibition of these channels also radiosensitised GB cells grown
ectopically in mice during radiotherapy. The authors conclude that the
KCa3.1-mediated response is required for the survival of irradiated cells
[125]. The radiogenic activity of KCa3.1 has been reported in other cancer cell
lines [126]. Pharmacological targeting of KCa3.1 interferes with cell cycle
control, involving CamKII, a long-acting signaling mediator. CamKII inhibits
cdc25 phosphatase, which prevents the activation of phosphorylated cdc2.
Instead, the cyclin B-cdc2 complex, when activated, arrests the cell cycle in
G2/M [127]. As TRAM-34 had a negligible effect on the cell cycle of
unirradiated T98G cells, the above results are in agreement with Abdullaev et
al. (2010). The presence and functional significance of KCa3.1 on gliomas, regarding
proliferation as well as migration and invasion is also reviewed by Elias et al.
[128].
KCa3.1 contribution to membrane potential changes and to cell cycle signaling by promoting Ca2+ influx
During the cell cycle advance, the membrane potential is driven by the activity of potassium channels [129]. More negative Vm or hyperpolarization is necessary to pass through the G1/S transition, while less negative Vm precedes G2/M advance, and hence depolarization is necessary for mitosis entry [130, 5, 131]. Progression through checkpoints of the cell cycle is associated also with changes in potassium channel activity and Vm [9]. Regulation of ion flux is one of the Vm effects upon cellular signaling, as part of the mechanism influencing the cell cycle. Studies with cancerous cells show that the activity of KCa3.1 promotes Ca2+ influx by hyperpolarizing the cell membrane, which increases the driving force for Ca2+ influx ( Fig. 2, C ), promoting proliferation [23, 111, 132]. Intracellular Ca2+ regulation allows appropriate cell cycle progression. In particular, nucleoplasmic Ca2+ regulates cell proliferation by activation of tyrosine kinase receptors (RTKs; Fig. 2, C-B ), with subsequent translocation to the nucleus, where they are part of nuclear Ca2+ signaling. Finally, nuclear Ca2+ regulates the expression of genes involved in cell proliferation [133]. Cancer cells have different ways of regulating calcium signaling to proliferate and survive than normal cells [134]. Some transformed cells may proliferate at external Ca2+ concentrations as low as tens of micromolar [135], whereas normal cell proliferation appears to be more dependent on calcium influx from the external environment [136]. Nevertheless, some plasma membrane calcium channels are overexpressed or more active in cancer cells [137].
Cell cycle phases are characterized by Ca2+ transients activating multiple Ca2+ effectors downstream of the initial Ca2+ signal [138, 19]. Among those effectors that dynamically regulate Ca2+ entry are the Ca2+-sensitive potassium channels (KCa), which drive intracellular Ca2+ levels [16]. In various cell types, fluctuations in intracellular calcium concentration [Ca2+]i determine cell cycle progression during the early G1 phase, G1/S, and G2/M transitions [138, 137, 19]. During the G1/S transition, hyperpolarization can increment the driving force for calcium entry through cooperation between potassium and calcium channels ( Fig. 2, C ). For example, KCa3.1 up-regulation in colorectal cancer cells HCT116 contributes with an electrochemical driving force to calcium entry mechanisms [27]. Calcium entry is recognized to be mediated either by Store Operated Calcium Entry (SOCE) or Store Independent Calcium Entry (SICE) in breast cancer cells [139], as well as in other cancer cells, in which calcium channels change their expression in plasma membrane along the cell cycle. Nevertheless, in transformed or cancer cells the causal relationship between proliferation and calcium entry is not generalized in all cell types and physiological conditions. Possible new functions for calcium channels besides calcium influx remain to elucidate: p.e. enzymatic activity [140]. Likewise, non-conducting functions promoting cell proliferation are also found in potassium channels, either voltage-dependent Kv1.3 or calcium-dependent KCa3.1 [141, 142]. Calcium-activated channels also appear to form complexes with Ca2+ channels. Thus, they functionally cooperate [16], as will be discussed here.
KCa3.1 promotes proliferation by its cooperation with Ca2+ channels
Ca2+ influx is essential for MCF-7 cell proliferation since it
can be suppressed by a decrease in the extracellular Ca2+
concentration [Ca2+]O [143]. In non-excitable cells, KCa and Ca2+ channels make a low-energy cost-positive feedback loop in Ca2+ Ca2+ channel complexes found in cancer cells contribute to
cancer-associated functions such as cell proliferation, among other
hallmark-related phenomena, allowing a fine regulation of [Ca2+]i [16]. Given the relatively high affinity of the
channel (KdCa: 188 nM) [40] and the fact that the basal [Ca2+]i is close to 100 nM [144], a change carrying a
new level of up to 200 nM may well activate the channel by more than half of
its operating capacity. For the reasons above and the small diffusion distances
of the cell, this proximity is not necessary to activate the channel. This
feature may reflect the importance of fine local regulation of the channel and
the [Ca2+]i
signal for cellular processes, e.g. proliferation.
Co-expression of KCa3.1 and the SOCE participant channel
Orai1 are detected nearby in microglia [145] or evidenced by
co-immunoprecipitation upon overexpression in HEK 293 cells [146].
Correspondingly, SOCE is important for normal or pathological cellular
processes, also regulated by KCa3.1 activity, as in Colorectal cancer (CRC)
[27] and neutrophils [147]. For instance, proliferation is promoted by
KCa-activation including KCa3.1, which cooperates to sustain Ca2+ entry mainly through SOCE in normal cells such as T-Cells
[148], cardiac progenitor cells (eCPCs), bone marrow (BM)-derived MSCs [149],
and in a cancerous chondrocyte cell line (OUMS-27), in response to histamine
release (Fig. 2, interaction between A, B and C) [150]. The cooperation between
KCa3.1-mediated hyperpolarization and TRP channels produces the proliferating
signal, e.g., in MCF-7, TRPC1 provides Ca2+ entry, which regulates cell proliferation via ERK1/2
phosphorylation (Fig. 2, pathway C-E-G) [151], cooperating functionally with
KCa3.1 and regulating cell cycle progression in G1 phase and G1/S transition.
Expression of both channels increases at the end of the G1 phase, and silencing
of either KCa3.1 or TRPC1 induces G0/G1 cell cycle arrest [93]. In lung
carcinoma NSCLC cell line, the TRPC1 participates in G1/S transition by
mediating entry, which is
crucial to EGFR signaling regulation involving cyclin D1 and D3 expression, as
well as MAPK downstream pathways [99]. In this case, further study is needed to
know whether KCa3.1 cooperates with TRPC1. However, the presence of these two
channels in those lung tissue cancers is striking. On the other hand, in
prostate cancer cell line LNCaP there is evidence of cooperation and
co-expression with the TRPV6 channel. This channel has been proposed as a
relevant means of passive Ca2+ influx in response to hyperpolarization associated with
KCa3.1 activity. Besides, immunoprecipitation experiments indicated a close
physical interaction between the KCa3.1 and TRPV6 in these cells, and that
KCa3.1 channels are up-regulated in prostate cancer (PCa) cells vs non-PCa
tissues [111], suggesting specific cancer calcium signaling alterations that
promote entry through
TRPV6. Also, TRPV4 regulates proliferation by co-activation with KCa3.1, in the
following tissues: human melanoma cell lines [152], reactive astrogliosis, a
pathologic cellular condition characterized by deregulated proliferation of
astrocytes [153], and a proliferative phenotype of human bronchial smooth
muscle (HBSM) [154]. Finally, co-activation was also found in Daudi lymphoma
cells, where the intermediate conductance KCa activity depends on Ca2+Ca2+-permeable channel CD20. CD20 channel activation and KCa3.1
expression were promoted by a serum factor, which could be IGF-1, as the
authors suggested [100]. Then, a positive feedback mechanism is common among
cancer cells through the functional cooperation between KCa and Ca2+ channels. There is much evidence of its close localization
and physical interaction [155, 16, 93], although its proximity is not necessary
to activate the channel due to its relatively high affinity. Moreover, another
crucial physical intermolecular association in proliferative processes
involving KCa3.1 is the non-canonical functionality, discussed below.
Non-canonical role of KCa3.1 channel involved in cell cycle signaling
Recombinant human KCa3.1 channels induce cell proliferation in human embryonic kidney 293 cells (HEK293), independently of its ion-conducting capabilities or derived consequences. The increased proliferative effect of nonconducting pore mutant KCa3.1(GYG/AAA) in those cells was similar to that of the wild KCa3.1 version. Moreover, the channel does not need to be located at the plasma membrane (PM) in similar quantities as in the wild KCa3.1-expressing cells to keep its proliferative function, as the trafficking mutant expressing KCa3.1(L18A/L25A) mostly outside of PM also increases proliferation. This flux-independent proliferative influence may occur through signaling ERK1/2 and JNK pathways (Fig. 2, pathway: D(1)-E-G) [141].
Aside from soluble extracellular factors, there is also proliferative information coming from the stiffness of the extracellular matrix (EM) [156], which activates ERK and PI3K pathways [157]. The cross-talking between growth factor receptors and EM adhesive receptors, such as integrins, in the plasma membrane activates proliferative pathways, migration, and survival in cancer cells. The expression of integrins is also upregulated in tumor cells [158, 159]. In primary cultured alveolar cells, KCa3.1 and β-1 integrin interact physically and cooperate to regulate proliferation in the presence of fibronectin, an EM structural component (Fig. 2, pathway: D(2)-E-G). Besides KCa3.1, TRPC4 and β-1 integrin plasma membrane expression were stimulated by fibronectin during a wound-healing assay. KCa3.1 and TRPC4 channels have complementary roles in modulating the interaction of β-1 integrin with fibronectin during lung epithelial regeneration [Girault et al., 2015]. Although regeneration in the aforementioned epithelium is not a pathological process, this cooperation between channels involved in proliferating signals resembles the cases of cancer cell lines in the previous sections. Interestingly, adhesion to fibronectin, mediated by β1-integrin, also stimulates the formation of a macromolecular complex, also with Kv11.1(hERG1), and the ensuing current activation of the ion channel, in transfected cell lines from neuroblastoma (SH-SY5Y) and human embryonic kidney (293) [160]. Further studies on structure, function, up-regulation, and interaction between these ion channels and integrins may lead to some clues about the modulation of proliferative signaling networks, among other features relating to cancer hallmarks, and how these molecular interactions respond to extracellular signals; either diffusible, matrix related or a combination of both.
Conclusion
The KCa3.1 channel's electrophysiological or Vm-dependent and non-canonical capabilities are crucial in determining proliferation in many mitogen-dependent types of cancer. Maybe the relevance of each mechanism for proliferation depends upon the kind of cell expressing the channel and the environment to which the cell is exposed. KCa3.1 may involve basal or mitogen-regulated proliferation through Vm-dependent, non-canonical mechanisms, or both. Studies analyzing the cellular signaling pathway determining cancerous proliferation mechanisms may help to improve therapy approaches.
It is also important to identify the unspecific targets of some KCa3.1 inhibitors. In addition to its apoptotic function, mtKCa3.1 also plays a significant role in cellular energetics. Therefore, the use of membrane-permeable KCa3.1 inhibitors may have adjuvant effects in some cells by acting on both the plasma and mitochondrial membranes. Targeting KCa3.1 is a promising avenue for future cancer therapies, as it may impact more cancer hallmarks than just the related to proliferation.
Acknowledgements
This manuscript was supported by the institutions where the authors work: Universidad Central de Venezuela and Fundación Instituto de Estudios Avanzados.
We thank Professor Andrés Carmona for his assistance in correcting the language issues in the final manuscript.
We thank Ms. Andira Anzola for her kind assistance in proofreading the language of the original manuscript.
Author Contributions
Christian Calderón has made the composition of all the contents. Francisco Arvelo has improved all the contents and reviewed the references used.
Funding sources
This manuscript was supported economically by the authors.
Statement of ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors have not Disclosure Statement to declare.
References
| 1 | Arvelo F, Sojo F, Cotte C: Tumour progression and metastasis. Ecancermedicalscience 2016;10:617.
https://doi.org/10.3332/ecancer.2016.617 |
| 2 | Prevarskaya N, Skryma R, Shuba Y: Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol Rev 2018;98:559-621.
https://doi.org/10.1152/physrev.00044.2016 |
| 3 | Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011;144:646-674.
https://doi.org/10.1016/j.cell.2011.02.013 |
| 4 | Prevarskaya N, Skryma R, Shuba Y: Ion channels and the hallmarks of cancer. Trends in molecular medicine 2010;16:107-121.
https://doi.org/10.1016/j.molmed.2010.01.005 |
| 5 | Urrego D, Tomczak AP, Zahed F, Stühmer W, Pardo LA: Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci 2014;369:20130094.
https://doi.org/10.1098/rstb.2013.0094 |
| 6 | Comes N, Serrano-Albarrás A, Capera J, Serrano-Novillo C, Condom E, Ramón Y Cajal S, Ferreres JC, Felipe A: Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim Biophys Acta 2015;1848:2477-2492.
https://doi.org/10.1016/j.bbamem.2014.12.008 |
| 7 | Yang M, Brackenbury WJ: Membrane potential and cancer progression. Front Physiol 2013;4:185.
https://doi.org/10.3389/fphys.2013.00185 |
| 8 | Sizemore G, McLaughlin S, Newman M, Brundage K, Ammer A, Martin K, Pugacheva E, Coad J, Mattes MD, Yu H-G: Opening large-conductance potassium channels selectively induced cell death of triple-negative breast cancer. BMC Cancer 2020;20:595.
https://doi.org/10.1186/s12885-020-07071-1 |
| 9 | Huang X, Jan LY: Targeting potassium channels in cancer. J Cell Biol 2014;206:151-162.
https://doi.org/10.1083/jcb.201404136 |
| 10 | Serrano-Novillo C, Capera J, Colomer-Molera M, Condom E, Ferreres JC, Felipe A: Implication of Voltage-Gated Potassium Channels in Neoplastic Cell Proliferation. Cancers (Basel) 2019;11:E287.
https://doi.org/10.3390/cancers11030287 |
| 11 | Pei L, Wiser O, Slavin A, Mu D, Powers S, Jan LY, Hoey T: Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc Natl Acad Sci U S A 2003;100:7803-7807.
https://doi.org/10.1073/pnas.1232448100 |
| 12 | Leithner K, Hirschmugl B, Li Y, Tang B, Papp R, Nagaraj C, Stacher E, Stiegler P, Lindenmann J, Olschewski A, Olschewski H, Hrzenjak A: TASK-1 Regulates Apoptosis and Proliferation in a Subset of Non-Small Cell Lung Cancers. PLoS One 2016;11:e0157453.
https://doi.org/10.1371/journal.pone.0157453 |
| 13 | Mohr CJ, Steudel FA, Gross D, Ruth P, Lo W-Y, Hoppe R, Schroth W, Brauch H, Huber SM, Lukowski R: Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1 Cancers (Basel) 2019;11.
https://doi.org/10.3390/cancers11010109 |
| 14 | Ohya S, Kito H, Kajikuri J: [Ca2+-activated K+ channels as cancer therapeutic targets]. Nihon Yakurigaku Zasshi 2019;154:108-113.
https://doi.org/10.1254/fpj.154.108 |
| 15 | Kahl CR, Means AR: Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr Rev 2003;24:719-736.
https://doi.org/10.1210/er.2003-0008 |
| 16 | Guéguinou M, Chantôme A, Fromont G, Bougnoux P, Vandier C, Potier-Cartereau M: KCa and Ca(2+) channels: the complex thought. Biochim Biophys Acta 2014;1843:2322-2333.
https://doi.org/10.1016/j.bbamcr.2014.02.019 |
| 17 | Prevarskaya N, Ouadid-Ahidouch H, Skryma R, Shuba Y: Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol Sci 2014;369:20130097.
https://doi.org/10.1098/rstb.2013.0097 |
| 18 | Humeau J, Bravo-San Pedro JM, Vitale I, Nuñez L, Villalobos C, Kroemer G, Senovilla L: Calcium signaling and cell cycle: Progression or death. Cell Calcium 2018;70:3-15.
https://doi.org/10.1016/j.ceca.2017.07.006 |
| 19 | Patergnani S, Danese A, Bouhamida E, Aguiari G, Previati M, Pinton P, Giorgi C: Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int J Mol Sci 2020;21:E8323.
https://doi.org/10.3390/ijms21218323 |
| 20 | Toro L, Li M, Zhang Z, Singh H, Wu Y, Stefani E: MaxiK channel and cell signalling. Pflugers Arch 2014;466:875-886.
https://doi.org/10.1007/s00424-013-1359-0 |
| 21 | Jäger H, Dreker T, Buck A, Giehl K, Gress T, Grissmer S: Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol Pharmacol 2004;65:630-638.
https://doi.org/10.1124/mol.65.3.630 |
| 22 | Zhang Y, Feng Y, Chen L, Zhu J: Effects of Intermediate-Conductance Ca(2+)-Activated K(+) Channels on Human Endometrial Carcinoma Cells. Cell Biochem Biophys 2015;72:515-525.
https://doi.org/10.1007/s12013-014-0497-0 |
| 23 | Ouadid-Ahidouch H, Roudbaraki M, Delcourt P, Ahidouch A, Joury N, Prevarskaya N: Functional and molecular identification of intermediate-conductance Ca(2+)-activated K(+) channels in breast cancer cells: association with cell cycle progression. Am J Physiol Cell Physiol 2004;287:C125-C134.
https://doi.org/10.1152/ajpcell.00488.2003 |
| 24 | Steudel FA, Mohr CJ, Stegen B, Nguyen HY, Barnert A, Steinle M, Beer-Hammer S, Koch P, Lo W-Y, Schroth W, Hoppe R, Brauch H, Ruth P, Huber SM, Lukowski R: SK4 channels modulate Ca2+ signalling and cell cycle progression in murine breast cancer. Mol Oncol 2017;11:1172-1188.
https://doi.org/10.1002/1878-0261.12087 |
| 25 | Parihar AS, Coghlan MJ, Gopalakrishnan M, Shieh C-C: Effects of intermediate-conductance Ca2+-activated K+ channel modulators on human prostate cancer cell proliferation. Eur J Pharmacol 2003;471:157-164.
https://doi.org/10.1016/S0014-2999(03)01825-9 |
| 26 | Pchelintseva E, Djamgoz MBA: Mesenchymal stem cell differentiation: Control by calcium-activated potassium channels. J Cell Physiol 2018;233:3755-3768.
https://doi.org/10.1002/jcp.26120 |
| 27 | Ibrahim S, Chaigne J, Dakik H, Fourbon Y, Corset L, Lecomte T, Raoul W, Guéguinou M: SK4 oncochannels regulate calcium entry and promote cell migration in KRAS-mutated colorectal cancer. Cell Calcium 2021;96:102384.
https://doi.org/10.1016/j.ceca.2021.102384 |
| 28 | Du Y, Song W, Chen J, Chen H, Xuan Z, Zhao L, Chen J, Jin C, Zhou M, Tuo B, Zhao Y, Zheng S, Song P: The potassium channel KCa3.1 promotes cell proliferation by activating SKP2 and metastasis through the EMT pathway in hepatocellular carcinoma. Int J Cancer 2019;145:503-516.
https://doi.org/10.1002/ijc.32121 |
| 29 | Gross D, Bischof H, Maier S, Sporbeck K, Birkenfeld AL, Malli R, Ruth P, Proikas-Cezanne T, Lukowski R: IKCa channels control breast cancer metabolism including AMPK-driven autophagy. Cell Death Dis 2022;13:902.
https://doi.org/10.1038/s41419-022-05329-z |
| 30 | Sevelsted Møller L, Fialla AD, Schierwagen R, Biagini M, Liedtke C, Laleman W, Klein S, Reul W, Koch Hansen L, Rabjerg M, Singh V, Surra J, Osada J, Reinehr R, de Muckadell OBS, Köhler R, Trebicka J: The calcium-activated potassium channel KCa3.1 is an important modulator of hepatic injury. Sci Rep 2016;6:28770.
https://doi.org/10.1038/srep28770 |
| 31 | Klumpp L, Sezgin EC, Skardelly M, Eckert F, Huber SM: KCa3.1 Channels and Glioblastoma: In vitro Studies. Curr Neuropharmacol 2018;16:627-635.
https://doi.org/10.2174/1570159X15666170808115821 |
| 32 | D'Alessandro G, Limatola C, Catalano M: Functional Roles of the Ca2+-activated K+ Channel, KCa3.1, in Brain Tumors. Curr Neuropharmacol 2018;16:636-643.
https://doi.org/10.2174/0929867324666170713103621 |
| 33 | Michelucci A, Sforna L, Di Battista A, Franciolini F, Catacuzzeno L: Ca2+ -activated K+ channels regulate cell volume in human glioblastoma cells. J Cell Physiol 2023;238:2120-2134.
https://doi.org/10.1002/jcp.31072 |
| 34 | Faouzi M, Chopin V, Ahidouch A, Ouadid-Ahidouch H: Intermediate Ca2+-sensitive K+ channels are necessary for prolactin-induced proliferation in breast cancer cells. J Membr Biol 2010;234:47-56.
https://doi.org/10.1007/s00232-010-9238-5 |
| 35 | Zhao L-M, Su X-L, Wang Y, Li G-R, Deng X-L: KCa3.1 channels mediate the increase of cell migration and proliferation by advanced glycation endproducts in cultured rat vascular smooth muscle cells. Lab Invest 2013;93:159-167.
https://doi.org/10.1038/labinvest.2012.163 |
| 36 | Girault A, Chebli J, Privé A, Trinh NTN, Maillé E, Grygorczyk R, Brochiero E: Complementary roles of KCa3.1 channels and β1-integrin during alveolar epithelial repair. Respir Res 2015;16:100.
https://doi.org/10.1186/s12931-015-0263-x |
| 37 | Xu P, Mo X, Xia R, Jiang L, Zhang C, Xu H, Sun Q, Zhou G, Zhang Y, Wang Y, Xia H: KCNN4 promotes the progression of lung adenocarcinoma by activating the AKT and ERK signaling pathways. Cancer Biomark 2021;31:187-201.
https://doi.org/10.3233/CBM-201045 |
| 38 | Todesca LM, Maskri S, Brömmel K, Thale I, Wünsch B, Koch O, Schwab A: Targeting Kca3.1 Channels in Cancer. Cell Physiol Biochem 2021;55:131-144.
https://doi.org/10.33594/000000374 |
| 39 | Lee C-H, MacKinnon R: Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 2018;360:508-513.
https://doi.org/10.1126/science.aas9466 |
| 40 | Fioretti B, Castigli E, Micheli MR, Bova R, Sciaccaluga M, Harper A, Franciolini F, Catacuzzeno L: Expression and modulation of the intermediate- conductance Ca2+-activated K+ channel in glioblastoma GL-15 cells. Cell Physiol Biochem 2006;18:47-56.
https://doi.org/10.1159/000095135 |
| 41 | Vergara C, Latorre R, Marrion NV, Adelman JP: Calcium-activated potassium channels. Curr Opin Neurobiol 1998;8:321-329.
https://doi.org/10.1016/S0959-4388(98)80056-1 |
| 42 | Wong R, Schlichter LC: PKA reduces the rat and human KCa3.1 current, CaM binding, and Ca2+ signaling, which requires Ser332/334 in the CaM-binding C terminus. J Neurosci 2014;34:13371-13383.
https://doi.org/10.1523/JNEUROSCI.1008-14.2014 |
| 43 | Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele DJ, Hubbard SR, Skolnik EY: Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife 2016;5:e16093.
https://doi.org/10.7554/eLife.16093 |
| 44 | Klein H, Garneau L, Trinh NTN, Privé A, Dionne F, Goupil E, Thuringer D, Parent L, Brochiero E, Sauvé R: Inhibition of the KCa3.1 channels by AMP-activated protein kinase in human airway epithelial cells. Am J Physiol Cell Physiol 2009;296:C285-295.
https://doi.org/10.1152/ajpcell.00418.2008 |
| 45 | Sforna L, Megaro A, Pessia M, Franciolini F, Catacuzzeno L: Structure, Gating and Basic Functions of the Ca2+-activated K Channel of Intermediate Conductance. Curr Neuropharmacol 2018;16:608-617.
https://doi.org/10.2174/1570159X15666170830122402 |
| 46 | Ferreira R, Wong R, Schlichter LC: KCa3.1/IK1 Channel Regulation by cGMP-Dependent Protein Kinase (PKG) via Reactive Oxygen Species and CaMKII in Microglia: An Immune Modulating Feedback System? Front Immunol 2015;6:153.
https://doi.org/10.3389/fimmu.2015.00153 |
| 47 | Srivastava S, Choudhury P, Li Z, Liu G, Nadkarni V, Ko K, Coetzee WA, Skolnik EY: Phosphatidylinositol 3-Phosphate Indirectly Activates KCa3.1 via 14 Amino Acids in the Carboxy Terminus of KCa3.1 Mol Biol Cell 2006a;17:146-54.
https://doi.org/10.1091/mbc.e05-08-0763 |
| 48 | Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK, Yan Y, Backer JM, Unutmaz D, Coetzee WA, Skolnik EY: Histidine Phosphorylation of the Potassium Channel KCa3.1 by Nucleoside Diphosphate Kinase B Is Required for Activation of KCa3.1 and CD4 T Cells. Mol Cell 2006b;24:665-675.
https://doi.org/10.1016/j.molcel.2006.11.012 |
| 49 | Gerlach AC, Syme CA, Giltinan L, Adelman JP, Devor DC: ATP-dependent activation of the intermediate conductance, Ca2+-activated K+ channel, hIK1, is conferred by a C-terminal domain. J Biol Chem 2001;276:10963-10970.
https://doi.org/10.1074/jbc.M007716200 |
| 50 | Burg S, Shapiro S, Peretz A, Haimov E, Redko B, Yeheskel A, Simhaev L, Engel H, Raveh A, Ben-Bassat A, Murninkas M, Polak R, Haitin Y, Etzion Y, Attali B: Allosteric inhibitors targeting the calmodulin-PIP2 interface of SK4 K+ channels for atrial fibrillation treatment. Proc Natl Acad Sci U S A 2022;119:e2202926119.51 Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H, Skolnik EY: Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1 Proc Natl Acad Sci U S A 2008;105:14442-14446.
https://doi.org/10.1073/pnas.0803678105 |
| 52 | Srivastava S, Li Z, Lin L, Liu G, Ko K, Coetzee WA, Skolnik EY: The phosphatidylinositol 3-phosphate phosphatase myotubularin- related protein 6 (MTMR6) is a negative regulator of the Ca2+-activated K+ channel KCa3.1 Mol Cell Biol 2005;25:3630-3638.
https://doi.org/10.1128/MCB.25.9.3630-3638.2005 |
| 53 | Castle NA, London DO, Creech C, Fajloun Z, Stocker JW, Sabatier J-M: Maurotoxin: a potent inhibitor of intermediate conductance Ca2+-activated potassium channels. Mol Pharmacol 2003;63:409-418.
https://doi.org/10.1124/mol.63.2.409 |
| 54 | Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG: Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA 2000;97:8151-8156.
https://doi.org/10.1073/pnas.97.14.8151 |
| 55 | Agarwal JJ, Zhu Y, Zhang Q-Y, Mongin AA, Hough LB: TRAM-34, a putatively selective blocker of intermediate-conductance, calcium-activated potassium channels, inhibits cytochrome P450 activity. PLoS ONE 2013;8:e63028.
https://doi.org/10.1371/journal.pone.0063028 |
| 56 | Stocker JW, De Franceschi L, McNaughton-Smith GA, Corrocher R, Beuzard Y, Brugnara C: ICA-17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood 2003;101:2412-2418.
https://doi.org/10.1182/blood-2002-05-1433 |
| 57 | Devor DC, Singh AK, Frizzell RA, Bridges RJ: Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am J Physiol 1996;271:L775-784.
https://doi.org/10.1152/ajplung.1996.271.5.L775 |
| 58 | King B, Rizwan AP, Asmara H, Heath NC, Engbers JDT, Dykstra S, Bartoletti TM, Hameed S, Zamponi GW, Turner RW: IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep 2015;11:175-182.
https://doi.org/10.1016/j.celrep.2015.03.026 |
| 59 | Strøbaek D, Teuber L, Jørgensen TD, Ahring PK, Kjaer K, Hansen RS, Olesen SP, Christophersen P, Skaaning-Jensen B: Activation of human IK and SK Ca2+ -activated K+ channels by NS309 (6, 7-dichloro-1H-indole-2, 3-dione 3-oxime). Biochim Biophys Acta 2004;1665:1-5.
https://doi.org/10.1016/j.bbamem.2004.07.006 |
| 60 | Morimura K, Yamamura H, Ohya S, Imaizumi Y: Voltage-dependent Ca2+-channel block by openers of intermediate and small conductance Ca2+-activated K+ channels in urinary bladder smooth muscle cells. J Pharmacol Sci 2006;100:237-241.
https://doi.org/10.1254/jphs.SC0060011 |
| 61 | Sankaranarayanan A, Raman G, Busch C, Schultz T, Zimin PI, Hoyer J, Köhler R, Wulff H: Naphtho [1, 2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol 2009;75:281-295.
https://doi.org/10.1124/mol.108.051425 |
| 62 | Coleman N, Brown BM, Oliván-Viguera A, Singh V, Olmstead MM, Valero MS, Köhler R, Wulff H: New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3.1 Mol Pharmacol 2014;86:342-357.
https://doi.org/10.1124/mol.114.093286 |
| 63 | Joiner WJ, Khanna R, Schlichter LC, Kaczmarek LK: Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J Biol Chem 2001;276:37980-37985.
https://doi.org/10.1074/jbc.M104965200 |
| 64 | Syme CA, Hamilton KL, Jones HM, Gerlach AC, Giltinan L, Papworth GD, Watkins SC, Bradbury NA, Devor DC: Trafficking of the Ca2+-activated K+ channel, hIK1, is dependent upon a C-terminal leucine zipper. J Biol Chem 2003;278:8476-8486.
https://doi.org/10.1074/jbc.M210072200 |
| 65 | Jones HM, Hamilton KL, Papworth GD, Syme CA, Watkins SC, Bradbury NA, Devor DC: Role of the NH2 terminus in the assembly and trafficking of the intermediate conductance Ca2+-activated K+ channel hIK1 J Biol Chem 2004;279:15531-15540.
https://doi.org/10.1074/jbc.M400069200 |
| 66 | Jones HM, Hamilton KL, Devor DC: Role of an S4-S5 linker lysine in the trafficking of the Ca(2+)-activated K(+) channels IK1 and SK3 J Biol Chem 2005;280:37257-37265.
https://doi.org/10.1074/jbc.M508601200 |
| 67 | Schwab A, Nechyporuk-Zloy V, Gassner B, Schulz C, Kessler W, Mally S, Römer M, Stock C: Dynamic redistribution of calcium sensitive potassium channels (hK(Ca)3.1) in migrating cells. J Cell Physiol 2012;227:686-696.
https://doi.org/10.1002/jcp.22776 |
| 68 | Balut CM, Loch CM, Devor DC: Role of ubiquitylation and USP8-dependent deubiquitylation in the endocytosis and lysosomal targeting of plasma membrane KCa3.1 FASEB J 2011;25:3938-3948.
https://doi.org/10.1096/fj.11-187005 |
| 69 | Balut CM, Gao Y, Murray SA, Thibodeau PH, Devor DC: ESCRT-dependent targeting of plasma membrane localized KCa3.1 to the lysosomes. Am J Physiol Cell Physiol 2010;299:C1015-1027.
https://doi.org/10.1152/ajpcell.00120.2010 |
| 70 | Catacuzzeno L, Fioretti B, Franciolini F: Expression and Role of the Intermediate-Conductance Calcium-Activated Potassium Channel KCa3.1 in Glioblastoma. J Signal Transduct 2012;2012:421564.
https://doi.org/10.1155/2012/421564 |
| 71 | Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG: Up-regulation of the IKCa1 Potassium Channel during T-cell Activation: MOLECULAR MECHANISM AND FUNCTIONAL CONSEQUENCES *. Journal of Biological Chemistry 2000;275:37137-37149.
https://doi.org/10.1074/jbc.M003941200 |
| 72 | Cheong A, Bingham AJ, Li J, Kumar B, Sukumar P, Munsch C, Buckley NJ, Neylon CB, Porter KE, Beech DJ, Wood IC: Downregulated REST Transcription Factor Is a Switch Enabling Critical Potassium Channel Expression and Cell Proliferation. Molecular Cell 2005;20:45-52.
https://doi.org/10.1016/j.molcel.2005.08.030 |
| 73 | Pardo LA, Stühmer W: The roles of K(+) channels in cancer. Nat Rev Cancer 2014;14:39-48.
https://doi.org/10.1038/nrc3635 |
| 74 | Catacuzzeno L, Sforna L, Esposito V, Limatola C, Franciolini F: Ion Channels in Glioma Malignancy. Rev Physiol Biochem Pharmacol 2021;181:223-267.
https://doi.org/10.1007/112_2020_44 |
| 75 | Park S, Kim JA, Joo KY, Choi S, Choi E-N, Shin J-A, Han K-H, Jung S-C, Suh SH: Globotriaosylceramide leads to K(Ca)3.1 channel dysfunction: a new insight into endothelial dysfunction in Fabry disease. Cardiovasc Res 2011;89:290-299.
https://doi.org/10.1093/cvr/cvq333 |
| 76 | Thurber AE, Nelson M, Frost CL, Levin M, Brackenbury WJ, Kaplan DL: IK channel activation increases tumor growth and induces differential behavioral responses in two breast epithelial cell lines. Oncotarget 2017;8:42382-42397.
https://doi.org/10.18632/oncotarget.16389 |
| 77 | Bianchi L, Wible B, Arcangeli A, Taglialatela M, Morra F, Castaldo P, Crociani O, Rosati B, Faravelli L, Olivotto M, Wanke E: herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells? Cancer Res 1998;58:815-822.
|
| 78 | Bulk E, Ay A-S, Hammadi M, Ouadid-Ahidouch H, Schelhaas S, Hascher A, Rohde C, Thoennissen NH, Wiewrodt R, Schmidt E, Marra A, Hillejan L, Jacobs AH, Klein H-U, Dugas M, Berdel WE, Müller-Tidow C, Schwab A: Epigenetic dysregulation of KCa 3.1 channels induces poor prognosis in lung cancer. Int J Cancer 2015;137:1306-1317.
https://doi.org/10.1002/ijc.29490 |
| 79 | Liu Y, Lv H, Wu X, Zhou J, Shi Y, Wen J: Demethylation of Repressor Element-1 Silencing Transcription (REST) Suppresses the Malignant Phenotype of Breast Cancer via MMP9 Oncol Res 2017b;25:445-454.
https://doi.org/10.3727/096504016X14747368729786 |
| 80 | Ohya S, Kanatsuka S, Hatano N, Kito H, Matsui A, Fujimoto M, Matsuba S, Niwa S, Zhan P, Suzuki T, Muraki K: Downregulation of the Ca(2+)-activated K(+) channel KC a3.1 by histone deacetylase inhibition in human breast cancer cells. Pharmacol Res Perspect 2016a;4:e00228.
https://doi.org/10.1002/prp2.228 |
| 81 | Cheong A, Wood IC, Beech DJ: Less REST, more vascular disease? Regulation of cell cycle and migration of vascular smooth muscle cells. Cell Cycle 2006;5:129-131.
https://doi.org/10.4161/cc.5.2.2310 |
| 82 | Cheong A, Wood IC, Beech DJ: Less REST, more vascular disease? Regulation of cell cycle and migration of vascular smooth muscle cells. Cell Cycle 2006;5:129-131.
https://doi.org/10.4161/cc.5.2.2310 |
| 83 | Negrini S, Prada I, D'Alessandro R, Meldolesi J: REST: an oncogene or a tumor suppressor? Trends Cell Biol 2013;23:289-295.
https://doi.org/10.1016/j.tcb.2013.01.006 |
| 84 | Liu L, Zhan P, Nie D, Fan L, Lin H, Gao L, Mao X: Intermediate-Conductance-Ca2-Activated K Channel Intermediate-Conductance Calcium-Activated Potassium Channel (IKCa1) is Upregulated and Promotes Cell Proliferation in Cervical Cancer. Med Sci Monit Basic Res 2017a;23:45-57.
https://doi.org/10.12659/MSMBR.901462 |
| 85 | Koegel H, Kaesler S, Burgstahler R, Werner S, Alzheimer C: Unexpected down-regulation of the hIK1 Ca2+-activated K+ channel by its opener 1-ethyl-2-benzimidazolinone in HaCaT keratinocytes. Inverse effects on cell growth and proliferation. J Biol Chem 2003;278:3323-3330.
https://doi.org/10.1074/jbc.M208914200 |
| 86 | Enomoto K, Cossu MF, Maeno T, Edwards C, Oka T: Involvement of the Ca2+-dependent K+ channel activity in the hyperpolarizing response induced by epidermal growth factor in mammary epithelial cells. FEBS Lett 1986;203:181-184.
https://doi.org/10.1016/0014-5793(86)80738-4 |
| 87 | Strobl JS, Wonderlin WF, Flynn DC: Mitogenic signal transduction in human breast cancer cells. Gen Pharmacol 1995;26:1643-1649.
https://doi.org/10.1016/0306-3623(95)00062-3 |
| 88 | Wegman EA, Young JA, Cook DI: A 23-pS Ca2(+)-activated K+ channel in MCF-7 human breast carcinoma cells: an apparent correlation of channel incidence with the rate of cell proliferation. Pflugers Arch 1991;417:562-570.
https://doi.org/10.1007/BF00372952 |
| 89 | Borowiec A-S, Hague F, Harir N, Guénin S, Guerineau F, Gouilleux F, Roudbaraki M, Lassoued K, Ouadid-Ahidouch H: IGF-1 activates hEAG K(+) channels through an Akt-dependent signaling pathway in breast cancer cells: role in cell proliferation. J Cell Physiol 2007;212:690-701.
https://doi.org/10.1002/jcp.21065 |
| 90 | Roy JW, Cowley EA, Blay J, Linsdell P: The intermediate conductance Ca2+-activated K+ channel inhibitor TRAM-34 stimulates proliferation of breast cancer cells via activation of oestrogen receptors. Br J Pharmacol 2010;159:650-658.
https://doi.org/10.1111/j.1476-5381.2009.00557.x |
| 91 | Brooks SC, Locke ER, Soule HD: Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J Biol Chem 1973;248:6251-6253.
https://doi.org/10.1016/S0021-9258(19)43537-0 |
| 92 | Brooks SC, Skafar DF: From ligand structure to biological activity: modified estratrienes and their estrogenic and antiestrogenic effects in MCF-7 cells. Steroids 2004;69:401-418.
https://doi.org/10.1016/j.steroids.2004.03.014 |
| 93 | Faouzi M, Hague F, Geerts D, Ay A-S, Potier-Cartereau M, Ahidouch A, Ouadid-Ahidouch H: Functional cooperation between KCa3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016;7:36419-36435.
https://doi.org/10.18632/oncotarget.9261 |
| 94 | Brockman JL, Schroeder MD, Schuler LA: PRL activates the cyclin D1 promoter via the Jak2/Stat pathway. Mol Endocrinol 2002;16:774-784.
https://doi.org/10.1210/mend.16.4.0817 |
| 95 | Tworoger SS, Hankinson SE: Prolactin and breast cancer risk. Cancer Lett 2006;243:160-169.
https://doi.org/10.1016/j.canlet.2006.01.032 |
| 96 | Schroeder MD, Symowicz J, Schuler LA: PRL modulates cell cycle regulators in mammary tumor epithelial cells. Mol Endocrinol 2002;16:45-57.
https://doi.org/10.1210/mend.16.1.0762 |
| 97 | Shepherd MC, Duffy SM, Harris T, Cruse G, Schuliga M, Brightling CE, Neylon CB, Bradding P, Stewart AG: KCa3.1 Ca2+ activated K+ channels regulate human airway smooth muscle proliferation. Am J Respir Cell Mol Biol 2007;37:525-531.
https://doi.org/10.1165/rcmb.2006-0358OC |
| 98 | Yu Z-H, Wang Y-X, Song Y, Lu H-Z, Hou L-N, Cui Y-Y, Chen H-Z: Up-regulation of KCa3.1 promotes human airway smooth muscle cell phenotypic modulation. Pharmacol Res 2013;77:30-38.
https://doi.org/10.1016/j.phrs.2013.09.002 |
| 99 | Tajeddine N, Gailly P: TRPC1 protein channel is major regulator of epidermal growth factor receptor signaling. J Biol Chem 2012;287:16146-16157.
https://doi.org/10.1074/jbc.M112.340034 |
| 100 | Wang ZH, Shen B, Yao HL, Jia YC, Ren J, Feng YJ, Wang YZ: Blockage of intermediate-conductance-Ca(2+) -activated K(+) channels inhibits progression of human endometrial cancer. Oncogene 2007;26:5107-5114.
https://doi.org/10.1038/sj.onc.1210308 |
| 101 | Bilanges B, Posor Y, Vanhaesebroeck B: PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol 2019;20:515-534.
https://doi.org/10.1038/s41580-019-0129-z |
| 102 | Liu SI, Chi CW, Lui WY, Mok KT, Wu CW, Wu SN: Correlation of hepatocyte growth factor-induced proliferation and calcium-activated potassium current in human gastric cancer cells. Biochim Biophys Acta 1998;1368:256-266.
https://doi.org/10.1016/S0005-2736(97)00183-1 |
| 103 | Nguyen PL, Cho J: Pathophysiological Roles of Histamine Receptors in Cancer Progression: Implications and Perspectives as Potential Molecular Targets. Biomolecules 2021;11:1232.
https://doi.org/10.3390/biom11081232 |
| 104 | Panula P, Lintunen M, Karlstedt K: Histamine in brain development and tumors. Semin Cancer Biol 2000;10:11-14.
https://doi.org/10.1006/scbi.2000.0302 |
| 105 | Lin J-J, Zhao T-Z, Cai W-K, Yang Y-X, Sun C, Zhang Z, Xu Y-Q, Chang T, Li Z-Y: Inhibition of histamine receptor 3 suppresses glioblastoma tumor growth, invasion, and epithelial-to-mesenchymal transition. Oncotarget 2015;6:17107-17120.
https://doi.org/10.18632/oncotarget.3672 |
| 106 | Fioretti B, Catacuzzeno L, Sforna L, Aiello F, Pagani F, Ragozzino D, Castigli E, Franciolini F: Histamine hyperpolarizes human glioblastoma cells by activating the intermediate-conductance Ca2+-activated K+ channel. Am J Physiol, Cell Physiol 2009;297:C102-110.
https://doi.org/10.1152/ajpcell.00354.2008 |
| 107 | Malumbres M: Cyclin-dependent kinases. Genome Biol 2014;15:122.
https://doi.org/10.1186/gb4184 |
| 108 | Mani S, Wang C, Wu K, Francis R, Pestell R: Cyclin-dependent kinase inhibitors: novel anticancer agents. Expert Opin Investig Drugs 2000;9:1849-1870.
https://doi.org/10.1517/13543784.9.8.1849 |
| 109 | Tao R, Lau C-P, Tse H-F, Li G-R: Regulation of cell proliferation by intermediate-conductance Ca2+-activated potassium and volume-sensitive chloride channels in mouse mesenchymal stem cells. Am J Physiol Cell Physiol 2008;295:C1409-1416.
https://doi.org/10.1152/ajpcell.00268.2008 |
| 110 | Chen Y, Kuang D, Zhao X, Chen D, Wang X, Yang Q, Wan J, Zhu Y, Wang Y, Zhang S, Wang Y, Tang Q, Masuzawa M, Wang G, Duan Y: miR-497-5p inhibits cell proliferation and invasion by targeting KCa3.1 in angiosarcoma. Oncotarget 2016;7:58148-58161.
https://doi.org/10.18632/oncotarget.11252 |
| 111 | Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackière F, Bidaux G, Urbain R, Gosset P, Delcourt P, Fleurisse L, Slomianny C, Dewailly E, Mauroy B, Bonnal JL, Skryma R, Prevarskaya N: Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 2009;28:1792-1806.
https://doi.org/10.1038/onc.2009.25 |
| 112 | El-Deiry WS: p21(WAF1) Mediates Cell-Cycle Inhibition, Relevant to Cancer Suppression and Therapy. Cancer Res 2016;76:5189-5191.
https://doi.org/10.1158/0008-5472.CAN-16-2055 |
| 113 | Karimian A, Ahmadi Y, Yousefi B: Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst) 2016;42:63-71.
https://doi.org/10.1016/j.dnarep.2016.04.008 |
| 114 | Cho Y-C, Park JE, Park BC, Kim J-H, Jeong DG, Park SG, Cho S: Cell cycle-dependent Cdc25C phosphatase determines cell survival by regulating apoptosis signal-regulating kinase 1 Cell Death Differ 2015;22:1605-1617.
https://doi.org/10.1038/cdd.2015.2 |
| 115 | Abdullaev IF, Rudkouskaya A, Mongin AA, Kuo Y-H: Calcium-activated potassium channels BK and IK1 are functionally expressed in human gliomas but do not regulate cell proliferation. PLoS One 2010;5:e12304.
https://doi.org/10.1371/journal.pone.0012304 |
| 116 | Gavrilova-Ruch O, Schönherr K, Gessner G, Schönherr R, Klapperstück T, Wohlrab W, Heinemann SH: Effects of imipramine on ion channels and proliferation of IGR1 melanoma cells. J Membr Biol 2002;188:137-149.
https://doi.org/10.1007/s00232-001-0181-3 |
| 117 | Zuccolini P, Barbieri R, Sbrana F, Picco C, Gavazzo P, Pusch M: IK Channel-Independent Effects of Clotrimazole and Senicapoc on Cancer Cells Viability and Migration. Int J Mol Sci 2023;24:16285.
https://doi.org/10.3390/ijms242216285 |
| 118 | Bulk E, Todesca LM, Bachmann M, Szabo I, Rieke M, Schwab A: Functional expression of mitochondrial KCa3.1 channels in non-small cell lung cancer cells. Pflugers Arch 2022;474:1147-1157.
https://doi.org/10.1007/s00424-022-02748-x |
| 119 | Kovalenko I, Glasauer A, Schöckel L, Sauter DRP, Ehrmann A, Sohler F, Hägebarth A, Novak I, Christian S: Identification of KCa3.1 Channel as a Novel Regulator of Oxidative Phosphorylation in a Subset of Pancreatic Carcinoma Cell Lines. PLoS One 2016;11:e0160658.
https://doi.org/10.1371/journal.pone.0160658 |
| 120 | De Marchi U, Sassi N, Fioretti B, Catacuzzeno L, Cereghetti GM, Szabò I, Zoratti M: Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009;45:509-516.
https://doi.org/10.1016/j.ceca.2009.03.014 |
| 121 | Quast S-A, Berger A, Buttstädt N, Friebel K, Schönherr R, Eberle J: General Sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS One 2012;7:e39290.
https://doi.org/10.1371/journal.pone.0039290 |
| 122 | Sassi N, De Marchi U, Fioretti B, Biasutto L, Gulbins E, Franciolini F, Szabò I, Zoratti M: An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKCa3.1 Biochim Biophys Acta 2010;1797:1260-1267.
https://doi.org/10.1016/j.bbabio.2009.12.015 |
| 123 | Bachmann M, Rossa A, Varanita T, Fioretti B, Biasutto L, Milenkovic S, Checchetto V, Peruzzo R, Ahmad SA, Patel SH, Lukowski R, Edwards MJ, Ceccarelli M, Gulbins E, Zoratti M, Mattarei A, Szabo I: Pharmacological targeting of the mitochondrial calcium-dependent potassium channel KCa3.1 triggers cell death and reduces tumor growth and metastasis in vivo. Cell Death Dis 2022b;13:1055.
https://doi.org/10.1038/s41419-022-05463-8 |
| 124 | Szabo I, Zoratti M, Biasutto L: Targeting mitochondrial ion channels for cancer therapy. Redox Biol 2021;42:101846.
https://doi.org/10.1016/j.redox.2020.101846 |
| 125 | Stegen B, Butz L, Klumpp L, Zips D, Dittmann K, Ruth P, Huber SM: Ca2+-Activated IK K+ Channel Blockade Radiosensitizes Glioblastoma Cells. Mol Cancer Res 2015;13:1283-1295.
https://doi.org/10.1158/1541-7786.MCR-15-0075 |
| 126 | Palme D, Misovic M, Schmid E, Klumpp D, Salih HR, Rudner J, Huber SM: Kv3.4 potassium channel-mediated electrosignaling controls cell cycle and survival of irradiated leukemia cells. Pflugers Arch 2013;465:1209-1221.
https://doi.org/10.1007/s00424-013-1249-5 |
| 127 | Roth B, Huber SM: Ion Transport and Radioresistance. Rev Physiol Biochem Pharmacol 2022;183:217-249.
https://doi.org/10.1007/112_2020_33 |
| 128 | Elias AF, Lin BC, Piggott BJ: Ion Channels in Gliomas-From Molecular Basis to Treatment. Int J Mol Sci 2023;24:2530.
https://doi.org/10.3390/ijms24032530 |
| 129 | Abdul Kadir L, Stacey M, Barrett-Jolley R: Emerging Roles of the Membrane Potential: Action Beyond the Action Potential. Front Physiol 2018;9:1661.
https://doi.org/10.3389/fphys.2018.01661 |
| 130 | Blackiston DJ, McLaughlin KA, Levin M: Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle 2009;8:3527-3536.
https://doi.org/10.4161/cc.8.21.9888 |
| 131 | Barghouth PG, Thiruvalluvan M, Oviedo NJ: Bioelectrical regulation of cell cycle and the planarian model system. Biochim Biophys Acta 2015;1848:2629-2637.
https://doi.org/10.1016/j.bbamem.2015.02.024 |
| 132 | Bi D, Toyama K, Lemaître V, Takai J, Fan F, Jenkins DP, Wulff H, Gutterman DD, Park F, Miura H: The intermediate conductance calcium-activated potassium channel KCa3.1 regulates vascular smooth muscle cell proliferation via controlling calcium-dependent signaling. J Biol Chem 2013;288:15843-15853.
https://doi.org/10.1074/jbc.M112.427187 |
| 133 | Resende RR, Andrade LM, Oliveira AG, Guimarães ES, Guatimosim S, Leite MF: Nucleoplasmic calcium signaling and cell proliferation: calcium signaling in the nucleus. Cell Commun Signal 2013;11:14.
https://doi.org/10.1186/1478-811X-11-14 |
| 134 | Roderick HL, Cook SJ: Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 2008;8:361-375.
https://doi.org/10.1038/nrc2374 |
| 135 | Capiod T: Cell proliferation, calcium influx and calcium channels. Biochimie 2011;93:2075-2079.
https://doi.org/10.1016/j.biochi.2011.07.015 |
| 136 | Capiod T: The need for calcium channels in cell proliferation. Recent Pat Anticancer Drug Discov 2013;8:4-17.
https://doi.org/10.2174/15748928130102 |
| 137 | Déliot N, Constantin B: Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochim Biophys Acta 2015;1848:2512-2522.
https://doi.org/10.1016/j.bbamem.2015.06.009 |
| 138 | Hodeify R, Yu F, Courjaret R, Nader N, Dib M, Sun L, Adap E, Hubrack S, Machaca K: Regulation and Role of Store-Operated Ca2+ Entry in Cellular Proliferation [Internet]; in Kozak JA, Putney JW (eds): Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL), CRC Press/Taylor & Francis, 2018, pp 215-240 [cited 2021 Apr 30].Available from: http://www.ncbi.nlm.nih.gov/books/NBK531436/
https://doi.org/10.1201/9781315152592-12 |
| 139 | Chamlali M, Rodat-Despoix L, Ouadid-Ahidouch H: Store-Independent Calcium Entry and Related Signaling Pathways in Breast Cancer. Genes (Basel) 2021;12:994.
https://doi.org/10.3390/genes12070994 |
| 140 | Borowiec A-S, Bidaux G, Pigat N, Goffin V, Bernichtein S, Capiod T: Calcium channels, external calcium concentration and cell proliferation. Eur J Pharmacol 2014;739:19-25.
https://doi.org/10.1016/j.ejphar.2013.10.072 |
| 141 | Millership JE, Devor DC, Hamilton KL, Balut CM, Bruce JIE, Fearon IM: Calcium-activated K+ channels increase cell proliferation independent of K+ conductance. Am J Physiol, Cell Physiol 2011;300:C792-802.
https://doi.org/10.1152/ajpcell.00274.2010 |
| 142 | Cidad P, Jiménez-Pérez L, García-Arribas D, Miguel-Velado E, Tajada S, Ruiz-McDavitt C, López-López JR, Pérez-García MT: Kv1.3 channels can modulate cell proliferation during phenotypic switch by an ion-flux independent mechanism. Arterioscler Thromb Vasc Biol 2012;32:1299-1307.
https://doi.org/10.1161/ATVBAHA.111.242727 |
| 143 | Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H: Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol 2011;226:542-551.
https://doi.org/10.1002/jcp.22363 |
| 144 | Marchi S, Pinton P: Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol 2016;29:1-6.
https://doi.org/10.1016/j.coph.2016.03.002 |
| 145 | Ferreira R, Schlichter LC: Selective activation of KCa3.1 and CRAC channels by P2Y2 receptors promotes Ca(2+) signaling, store refilling and migration of rat microglial cells. PLoS One 2013;8:e62345.
https://doi.org/10.1371/journal.pone.0062345 |
| 146 | Duffy SM, Ashmole I, Smallwood DT, Leyland ML, Bradding P: Orai/CRACM1 and KCa3.1 ion channels interact in the human lung mast cell plasma membrane. Cell Commun Signal 2015;13:32.
https://doi.org/10.1186/s12964-015-0112-z |
| 147 | Grimes D, Johnson R, Pashos M, Cummings C, Kang C, Sampedro GR, Tycksen E, McBride HJ, Sah R, Lowell CA, Clemens RA: ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc Natl Acad Sci U S A 2020;117:24403-24414.
https://doi.org/10.1073/pnas.2008032117 |
| 148 | Panyi G, Beeton C, Felipe A: Ion channels and anti-cancer immunity. Philos Trans R Soc Lond B Biol Sci 2014;369:20130106.
https://doi.org/10.1098/rstb.2013.0106 |
| 149 | Vigneault P, Naud P, Qi X, Xiao J, Villeneuve L, Davis DR, Nattel S: Calcium-dependent potassium channels control proliferation of cardiac progenitor cells and bone marrow-derived mesenchymal stem cells. J Physiol 2018;596:2359-2379.
https://doi.org/10.1113/JP275388 |
| 150 | Funabashi K, Ohya S, Yamamura H, Hatano N, Muraki K, Giles W, Imaizumi Y: Accelerated Ca2+ entry by membrane hyperpolarization due to Ca2+-activated K+ channel activation in response to histamine in chondrocytes. Am J Physiol Cell Physiol 2010;298:C786-797.
https://doi.org/10.1152/ajpcell.00469.2009 |
| 151 | El Hiani Y, Lehen'kyi V, Ouadid-Ahidouch H, Ahidouch A: Activation of the calcium-sensing receptor by high calcium induced breast cancer cell proliferation and TRPC1 cation channel over-expression potentially through EGFR pathways. Arch Biochem Biophys 2009;486:58-63.
https://doi.org/10.1016/j.abb.2009.03.010 |
| 152 | Olivan-Viguera A, Garcia-Otin AL, Lozano-Gerona J, Abarca-Lachen E, Garcia-Malinis AJ, Hamilton KL, Gilaberte Y, Pueyo E, Köhler R: Pharmacological activation of TRPV4 produces immediate cell damage and induction of apoptosis in human melanoma cells and HaCaT keratinocytes. PLoS One 2018;13:e0190307.
https://doi.org/10.1371/journal.pone.0190307 |
| 153 | Yi M, Wei T, Wang Y, Lu Q, Chen G, Gao X, Geller HM, Chen H, Yu Z: The potassium channel KCa3.1 constitutes a pharmacological target for astrogliosis associated with ischemia stroke. J Neuroinflammation 2017;14:203.
https://doi.org/10.1186/s12974-017-0973-8 |
| 154 | Yu Z, Wang Y, Qin L, Chen H: Functional Cooperation between KCa3.1 and TRPV4 Channels in Bronchial Smooth Muscle Cell Proliferation Associated with Chronic Asthma. Front Pharmacol 2017;8:559.
https://doi.org/10.3389/fphar.2017.00559 |
| 155 | Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F: Molecular interaction and functional coupling between type 3 inositol 1, 4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. Eur J Cancer 2013;49:3738-3751.
https://doi.org/10.1016/j.ejca.2013.07.013 |
| 156 | Seetharaman S, Etienne-Manneville S: Integrin diversity brings specificity in mechanotransduction. Biol Cell 2018;110:49-64.
https://doi.org/10.1111/boc.201700060 |
| 157 | Keely PJ: Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J Mammary Gland Biol Neoplasia 2011;16:205-219.
https://doi.org/10.1007/s10911-011-9226-0 |
| 158 | Marelli UK, Rechenmacher F, Sobahi TRA, Mas-Moruno C, Kessler H: Tumor Targeting via Integrin Ligands. Front Oncol 2013;3:222.
https://doi.org/10.3389/fonc.2013.00222 |
| 159 | Ellert-Miklaszewska A, Poleszak K, Pasierbinska M, Kaminska B: Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int J Mol Sci 2020;21:E888.
https://doi.org/10.3390/ijms21030888 |
| 160 | Cherubini A, Hofmann G, Pillozzi S, Guasti L, Crociani O, Cilia E, Di Stefano P, Degani S, Balzi M, Olivotto M, Wanke E, Becchetti A, Defilippi P, Wymore R, Arcangeli A: Human ether-a-go-go-related gene 1 channels are physically linked to beta1 integrins and modulate adhesion-dependent signaling. Mol Biol Cell 2005;16:2972-2983.
https://doi.org/10.1091/mbc.e04-10-0940 |