Original Article - DOI:10.33594/000000830
Accepted 13 October 2025 - Published online
18 November 2025
3-Acetyl-11-Keto-Beta-Boswellic Acid Inhibits Adipogenesis by Suppressing Autophagy and Inducing AMPK Phosphorylation in 3T3-L1 Cells
bDepartment of Pharmacy, Abdul Wali Khan University Mardan, Mardan 23200 Khyber Pakhtunkhwa, Pakistan,
cDepartment of Pharmacy, Korea University, Sejong 20019, South Korea,
dDepartment of Pharmaceutical Sciences, College of Pharmacy, Texas A & M University, College Station, Texas 77843, USA
Keywords
Abstract
Background/Aims:
Adipogenesis, a process involving preadipocyte differentiation and lipid droplet accumulation, is linked to obesity. 3-Acetyl-11-keto-beta-boswellic acid (AKBA), a frankincense-derived triterpene, has anti-inflammatory and cancer properties, but its role in regulating adipocyte differentiation and adipogenesis remains unclear.Methods:
3T3-L1 preadipocytes were induced to differentiate into adipocytes using a differentiation cocktail, with or without varying concentrations of AKBA. Cell viability was assessed using the MTT assay, while lipid accumulation was quantified through Oil Red O staining in the differentiated 3T3-L1 adipocytes. Apoptosis was determined using the FITC (fluorescein isothiocyanate) annexin V apoptosis detection kit. Expression of pro- and anti-apoptotic proteins Bax and Bcl2, adipogenic transcription factors, lipid accumulation-related proteins, and autophagy marker proteins were analyzed via Western blotting. Additionally, molecular docking simulations were conducted to investigate the binding affinity and interactions between the bioactive compounds and the targeted proteins.Results:
AKBA treatment inhibited adipocyte differentiation by suppressing the protein levels of essential adipogenic markers CCAAT/enhancer binding protein beta (C/EBPβ), CCAAT/enhancer binding protein alpha (C/EBPα), and peroxisome proliferator-activated receptor gamma (PPARγ). Further, AKBA treatment significantly reduced lipid accumulation and increased apoptosis in 3T3-L1 cells by increasing Bax/Bcl2 ratio. Notably, AKBA treatment profoundly reduced autophagy indicators, including ATG5 and LC3b protein expression in differentiated 3T3-L1 cells, concurrent with increased AMPK phosphorylation (P-AMPK). Further, a molecular docking study revealed that AKBA can block PPARγ and ATG5.Conclusion:
The results indicate that AKBA exhibits anti-adipogenesis effects by suppressing adipocyte-specific transcription factors C/EBPα and PPARγ, autophagy-related marker ATG5, promoting apoptosis and activating AMPK.Introduction
Obesity is a global health concern [1] marked by excessive or abnormal fat mass expansion and cellular lipid buildup resulting from an imbalance in energy intake and expenditure [2]. Obesity’s growing prevalence is especially concerning because it is a significant contributor to noncommunicable diseases like cancer, cardiovascular disease, and type 2 diabetes [3, 4]. Substantial evidence suggests that increased adipocyte number (hyperplasia) and size (hypertrophy) are responsible for adipose tissue expansion in the obese condition [2]. Hypertrophy is closely associated with adipogenesis, a process by which preadipocytes mature into adipocytes and accumulate lipids, to satisfy the increased lipid storage demand in obesity [2]. Consequently, adipogenesis is critical for regulating total fat mass [5].
Adipogenesis encompasses two phases: commitment and terminal differentiation. In the commitment phase, MSCs are converted into preadipocytes, which bear a morphological resemblance to their precursors but forfeit their capacity to differentiate into alternate cell lineages [6].. Preadipocytes undergo late differentiation to become adipocytes, which can produce lipids, are insulin-responsive, and manufacture proteins specific to adipocytes [6].
Adipogenesis is an essential process for fat cell formation. It involves various gene regulators, such as CCAAT/enhancer-binding proteins (C/EBPs) and peroxisome proliferator-activated receptor gamma (PPARγ) [7, 8]. These transcription factors can activate adipocyte-specific genes associated with lipogenesis and adipogenesis, such as lipoprotein lipase, adiponectin, fatty acid synthase, and perilipin [9]. Reducing these transcription factors' expression and activity has been suggested as an effective strategy for obesity treatment. Thus, comprehending the biological processes of adipose tissue growth and the essential elements involved in its regulation is critical for future obesity prevention and treatment therapies.
The therapeutic value of bioactive compounds for disease therapy has garnered significant interest in recent years. To date, many bioactive compounds have been isolated from natural sources and tested for their efficacy in different disease models [10]. Boswellic acids, a type of pentacyclic triterpene found in plants like Boswellia, are commonly used to treat ailments like arthritis, psoriasis, asthma, and cancers [11-13]. Previous research has indicated that boswellic acids are well tolerated and confer beneficial effects in humans [14]. For the past ten years, we have created a library of naturally isolated and synthetically modified natural products, including a collection of medicinally relevant triterpenes extracted from frankincense [15]. Of these bioactive substances, 3-Acetyl-11-keto-β-boswellic acid (AKBA) is the most potent against cancer [13, 15, 16]. Boswellic acids (BA) and AKBA lessened STZ-mediated production of cytokines that trigger inflammation in the circulation and CD3 cell recruitment to the pancreas. Additionally, AKBA was more effective in lowering STZ-mediated hyperglycemia [17, 18]. Boswellia serrata (predominantly containing Boswellic acids) was also found effective in preventing diet-induced obesity, hyperlipidemia, and insulin resistance. It was further demonstrated that Boswellia serrata inhibited food intake and pro-inflammatory cytokine production, along with increasing anti-inflammatory adiponectin production [19]. Boswellia serrata gum resin drastically lowered total, LDL, and triglyceride levels in diabetic patients' blood while raising HDL cholesterol [20]. Additionally, a combination of metformin and gum resin of Boswellia Serrata substantially reduced blood glucose, Hemoglobin A1c (HbA1C), insulin, LDL cholesterol, and triglycerides in diabetic patients compared to metformin treatment alone [21]. Despite these promising studies on the efficacy of Boswellic acids in obesity and diabetes, it is unclear whether it has any effect on adipocyte differentiation and lipid accumulation. More specifically, it examines the effects associated with adipogenesis and adipose tissue expansion, along with their underlying molecular and cellular mechanisms.
In the present investigation, we examined how AKBA affected variables linked to adipocyte development (C/EBPβ, C/EBPα, and PPARγ) and autophagy markers (LC3b and ATG5) in 3T3-L1 cells. Furthermore, AKBA’s role in lipid accumulation and AMPK phosphorylation was examined in adipocytes. Additionally, the potential of AKBA to block adipogenesis was validated through molecular docking of pharmacological targets.
Materials and Methods
Chemicals and reagents
Oil Red O staining solution, Dulbecco’s Modified Eagle
Medium (DMEM), MTT assay kit, Chemiluminescence substrate, Isobutyl methylxanthine (IBMX), insulin, and
dexamethasone were purchased from Sigma Aldrich (St. Louis, MO, USA). PPARγ (Cat. #2430S), C/EBPα (Cat.
#2295S),
C/EBPβ (Cat. #3082S), ATG5 (Cat. #2630S), P-AMPK (Thr172, Cat. #2531S), AMPK (Cat. #2532S), Bax (Cat.
#2772S),
Bcl2 (Cat. #2876S), and β-actin (Cat. #4967S) were obtained from Cell Signaling Technology, Danvers, MA,
USA.
FAS (Cat. #A0462) was purchased from Abclonal Technology (Woburn, MA, USA). LC3b (Cat. #AF4650) were
obtained
from Affinity Biosciences, Cincinnati, OH, USA. 3T3-L1 (Cat. #CL-173) preadipocytes were obtained from
ATCC
(Rockville, MD, USA) and maintained in DMEM supplemented with 10% bovine serum and 100 U/ml penicillin and
streptomycin.
Extraction and purification of AKBA
The oleo-gum resin of Boswellia sacra was gathered from multiple sites in the Dhofar governorate of
Oman
(2012) and certified by the herbarium at the Natural and Medicinal Sciences Research Center, University of
Nizwa, Oman (voucher specimen, BSHR-01/2020). After authentication of the sample, a 500 g sample of
air-dried
and crushed resin was extracted with distilled methanol (MeOH, 1.2 L) for 2-3 days with continuous and
vigorous
shaking after 30 min to get a yellowish residue (280 g). The MeOH extract (120 g) was analyzed using
column
chromatography (CC, SiO2) using a gradient polarity of n-hexane/ethyl acetate (EtOAc) with up to 100%
EtOAc,
yielding twelve fractions (BS1-12) as described before [22, 23]. Fractions 5-7 were combined after TLC
confirmed
the presence of AKBA and then loaded onto CC (600 g; 70-230 mesh) to isolate impure AKBA (800 mg with a
small
amount of KBA impurity) using 30-40% EtOAc/n-hexane. After fermenting with 0.6% ethanol, the resulting
sample
was transferred directly to a recycling chloroform HPLC. Purified AKBA (350 mg) was eluted at a flow rate
of 4
ml/min after a retention time of 42 minutes [22, 23].
Differentiation of preadipocytes into mature adipocytes
3T3-L1 preadipocytes were grown in DMEM with 10% FBS to initiate differentiation. After reaching
confluence two
days following plating (day 0), the cells were treated with differentiation stimulation medium containing
0.5mM
isobutylmethylxanthine (IBMX), 167 nM insulin, and 1mM dexamethasone for two days (day 2). The cells were
then
switched to a medium that contains DMEM with 167 nM insulin for another two days (till day 4). Next, the
cells
were grown in DMEM medium with 10% FBS for four more days (till day 8), as shown in Fig. 1. The cells were
subjected to specified treatments throughout the entire culture period (from day zero to day 8).
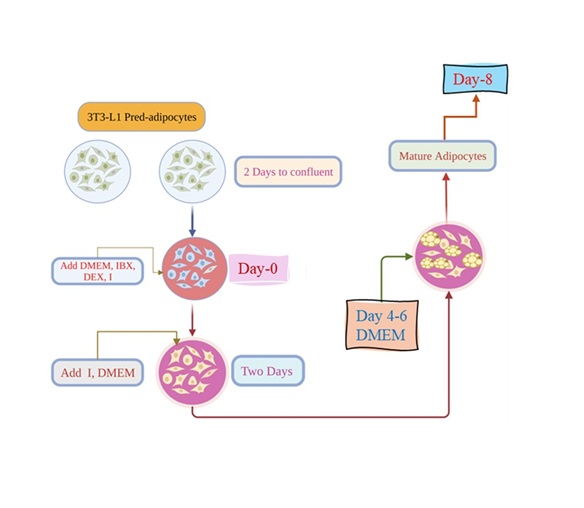
Fig. 1: rocedure for inducing differentiation of 3T3-L1 preadipocytes into mature adipocytes. DMEM; Dulbecco’s modified Eagle medium, IBX; isobutyl-methyl-xanthine, I; insulin, DEX; dexamethasone.
Cell viability assay
Cell viability was examined using the MTT test in 3T3-L1 cells treated with AKBA.
The cells were plated in a 96-well plate and cultured for 24 hours. Following treatment with varied
concentrations of AKBA for 24 hours, the media was removed, and cell viability was evaluated at 570 nm
with a
microplate reader (Bio-Rad, USA). The results displayed a percentage change in cell viability compared to
untreated control cells, determined via the following equation.
$$ \% \text{Viability} = \frac{\text{Absorbance of sample}}{\text{Absorbance of control}} \times 100 \hspace{50px} \text{(Eq - 1)}$$
Oil red O staining
3T3-L1 preadipocytes were treated with 3-isobutyl-1-methylxanthine, dexamethasone, and insulin (MDI) for 8
days
with and without AKBA (C-D). Undifferentiated 3T3-L1 cells without AKBA treatment were used as (C-N). The
influence of AKBA on adipocyte intracellular lipid accumulation was measured by oil red O (ORO) staining.
Adipocytes were fixed in 10% formalin for 1 hour, followed by permeabilization in 60% isopropanol for 5
minutes.
Fixed cells were stained with Oil Red O solution in 20% isopropanol for 1 hour, then washed four times
with H2O.
The lipid droplets were subsequently detected and photographed using an Evos XL Core (Invitrogen). To
estimate
the amount of neutral lipids, 100% isopropanol was used to extract Oil Red O from the cells, and the
optical
density absorbance at 510 nm was measured with a microplate reader (Bio-Rad, USA).
Preparation of cytosolic and nuclear fractions
The cytosolic fractions were prepared as previously reported [24]. Protein concentration was measured
using the
Bradford assay (Bio-Rad), with bovine serum albumin (BSA) as the standard, according to the manufacturer's
instruction.
Western blotting
Using a mix of a proteinase inhibitor cocktail (Roche) and RIPA buffer containing 50 mM Tris, pH 7.0, 150
mM
NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% NP-40, cells were lysed. 40µg of proteins were separated
using
12% SDS-PAGE gels and transferred to nitrocellulose membranes. Transferring gels to nitrocellulose
membranes was
achieved by electroblotting. The membranes were blocked with 5% nonfat milk in PBS-Tween buffer for 1 hour
and
then incubated overnight at 4°C with primary antibodies (C/EBPα, C/EBPβ, PPARγ, ATG5, LC3B, FAS,
Cell
Signaling
Technology, Danvers, Massachusetts, USA) at a 1:500 dilutions. The secondary antibody
(β-actin) was
then incubated at 1:5000 dilutions at room temperature for 1 hour. The ECL method (Thermo Scientific, USA)
was
used to detect protein signals, which were then visualized using the ChemiDoc System (Bio-Rad, USA).
Protein
expression was evaluated densitometrically using Image J software and normalized to β-actin standard bands
and
total AMPK for P-AMPK. Values are presented as percentage changes relative to the control group [25]. For
Bax
and Bcl2 (Cell Signaling Technology, Danvers, Massachusetts, USA) identification, 20μg of protein was
loaded per
well in a 15% SDS-PAGE gel and transferred on to PVDF membrane. The membrane was blocked
using Intercept®
(TBS) Blocking Buffer (Cat. #927-60001, LICORbio™, NE, USA) for 1h. Later, membrane was incubated
with
1:1000 primary antibody overnight at 4oC. Then, the membrane was incubated with 1:2000
secondary
antibody at RT for 1h. Fluorescence was imaged using Odyssey CLx Imager (LICORbio™, NE,
USA). Densitometry was performed using Image Studio™ 6.0 software, (LICORbio™, NE,
USA). Bax and
Bcl2 values were normalized to β-actin, and the fold change was used to represent the Bax/Bcl2 ratio.
Docking simulations
Crystal structures retrieval. The crystal structural data of the PPARγ Ligand Binding Domain
in
conjunction with the T0070907 inhibitor (PDB ID: 6C1I), and the inactivation of Autophagy protein 5 (ATG5)
by a
stapled peptide (PDB ID: 7W36), was obtained from the RCSB Protein Data Bank (PDB) server to analyze the
atomic
interactions of the AKBA with the protein's active pockets.
The Molecular Operating Environment (MOE) software, version 2022.02 [26], was employed to restore the lost
residues in the three-dimensional (3D) structures found in PPARγ and ATG5 proteins with the Loop
Modeler
algorithm and the Amber14:EHT forcefield [27, 28]. To add hydrogens that were missing and assign charges
at the
protein's terminal ends, the Quick Prep panel in MOE 2022.02 was used [26]. Additionally, the MOE software
was
used to add several structural deficiencies, such as missing atom types, angle definitions, forcefield
parameters, Van der Waals interactions, bond formations, and residue chirality.
Molecular docking. Molecular docking is a vital tool for in silico screening, which is increasingly
significant in rational drug design. Molecular docking simulates the interaction between a protein's
binding
site and a potential ligand, ensuring both energetic and geometric compatibility [29, 30]. We utilized the
Dock
function in MOE 2022.02 to conduct docking experiments between the AKBA compound and the PPARγ as
well as
ATG5
proteins [26]. Initially, we re-docked the native ligand in PPARγ to confirm the accuracy of our
docking
approach before examining the AKBA compounds. The validation of our docking protocol comprised of three
stages.
First, the ligand's geometry was optimized by rotating bonds to achieve proper alignment. Subsequently, we
used
the Triangle Matcher for ligand placement, generating 100 potential conformations. Each conformation's
binding
energy was calculated using the London dG Scoring function (Equation 2), incorporating factors such as
entropy
changes, flexibility loss, hydrogen bonding, metal interactions, and desolvation energies [26].
$$ G = c + E_{flex} + \sum_{\text{h-bonds}} c_{HB} f_{HB} + \sum_{\text{m-lig}} c_M f_M + \sum_{\text{atom i}} \Delta D_i \hspace{50px} \text{(Eq - 2)}$$
In the final stage, the best conformer was chosen from the initial 100 using the GBVI/WSA dG scoring
algorithm
in MOE 2022.02. The binding free energies of the top 30 conformations were further calculated using a
secondary
formula (Equation 3) that accounts for changes in entropy, electrostatic interactions, and van der Waals
forces
[26].
$$ \Delta G \approx c + a \left[ \frac{2}{3} (\Delta E_{coul} + \Delta E_{sol}) + \Delta E_{vdw} + \beta \Delta S A_{weighted} \right] \hspace{50px} \text{(Eq - 3)} $$
After validating our methodology by superimposing the native and re-docked ligands and assessing the Root
Mean
Square Deviation (RMSD), we proceeded to dock the AKBA with the PPARγ and ATG5 proteins’ active
sites. We
examined the relationships between every docked complex that was stored in PDB format. We employed the
Protein-Ligand Interaction Fingerprinting (PLIF) tool available in the MOE Database viewer to perform the
analysis.
Statistical analysis
The data is shown as mean ± Standard deviation (SD), with n = 3. Statistical analysis was performed using
one-way ANOVA with Tukey’s post hoc test or using an unpaired Student’s t-test. The statistical
significance of
each comparison was assessed based on P-values lower than 0.05.
Results
Effects of AKBA on 3T3-L1 cell viability
To evaluate the effect of AKBA on 3T3-L1 cell growth, 3T3-L1 cells were induced to differentiate into
adipocytes
over 8 days, after which the cytotoxicity of AKBA on the differentiated adipocytes was assessed [31]. The
3T3-L1
cells were administered with AKBA at dosages ranging from 0.78 µM to 100 µM, and their viability was
determined
using the MTT assay. AKBA at concentrations of 0.78, 1.56, and 3.1 µM had no cytotoxic effects on mature
adipocytes (Fig. 2). Treatment with 6.25 to 100 μM AKBA dramatically reduced mature adipocyte cell
viability by
15 to 60% compared to the control group (P<0.05, P<0.01, and P<0.001, Fig. 2). For subsequent
experiments, we used three AKBA concentrations: 2.5 µM (non-toxic to mature adipocytes) as well as 5 and
10 µM
(lower than IC50 that was determined at 53.2 µM in matured adipocytes).
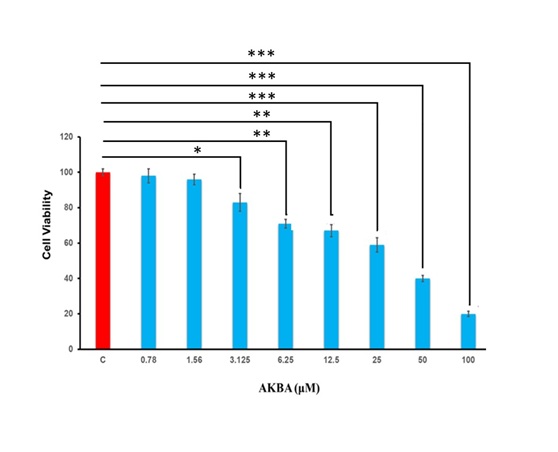
Fig. 2: AKBA reduced the viability of 3T3-L1 cells. The data is reported as mean ± standard deviation (SD) and presented as a percentage change relative to the control group. n=3. C; control. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test. *P<0.5, **P<0.01, ***P<0.001 vs. Control. P values < 0.05 were considered statistically significant for all comparisons.
AKBA suppressed lipid accumulation in 3T3-L1 cells
Adipocytes retain surplus lipids or glucose as triglycerides and are essential to control lipid metabolism
and
energy homeostasis by providing free fatty acids and glycerol when energy is required [32]. Thus, the
development of obesity is intimately linked to preadipocyte differentiation and increased lipid
accumulation
[33]. To determine the impact of AKBA on adipogenesis, the cytoplasmic lipid level was evaluated in mature
adipocytes. 3T3-L1 preadipocytes were administered with AKBA at various concentrations (2.5, 5, 10 µM)
during
the differentiation phase for 5 days (days 4–8). The intracellular lipid buildup was assessed by Oil Red O
(ORO)
staining (Fig. 3A and 3B). Compared to undifferentiated preadipocytes (C-N), mature adipocytes (C-D)
displayed a
considerable increase in lipid accumulation (Fig. 3A and 3B). Microscopy revealed that 2.5 µM, 5 µM, and
10 µM
AKBA significantly reduced ORO staining in differentiated 3T3-L1 adipocytes (Fig. 3A). Stained cells were
eluted
with isopropanol, and the cellular lipid was quantified spectrophotometrically at 510 nm using a
microplate
reader. We found that 2.5 µM, 5 µM, and 10 µM AKBA significantly reduced lipid buildup by 18%, 36%, and
60%,
respectively, in comparison to the control group (C-D) (P < 0.01, P<0.001, and P<0.001,
respectively,
Fig. 3B). The data suggest that AKBA decreased lipid formation in mature adipocytes.
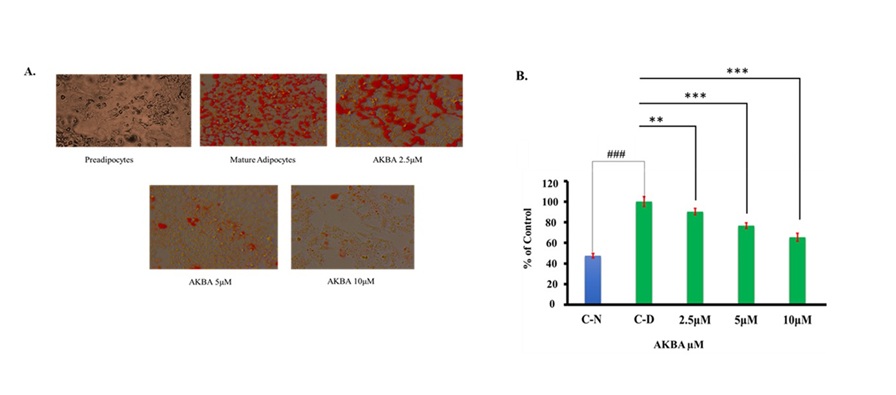
Fig. 3: AKBA suppressed lipid buildup in differentiated 3T3-L1 cells. (A) Representative images (40x magnification) of Oil Red O staining are shown. (B) Lipid quantification. Data are reported as mean ±SD, n=3. Statistical analysis was performed using an unpaired Student’s t-test; ###P<0.001 vs. C-N and one-way ANOVA with Tukey’s post hoc test; **P<0.01, ***P<0.001 vs. control group (C-D). P values < 0.05 were considered statistically significant for all comparisons.
AKBA promoted apoptosis in 3T3-L1 cells
The present study revealed that AKBA mediated a decrease in cell viability and lipid accumulation in
mature
adipocytes. Next, we assessed the impact of AKBA on apoptosis in mature adipocytes. We used Annexin V-FITC
and
PI double staining to examine the effect of AKBA on apoptosis in mature adipocytes. During apoptosis, the
membrane phospholipid phosphatidylserine translocates from the inner to the outer membrane, where it is
detected
by Annexin V-FITC, a phospholipid-binding protein [34]. Flow cytometry was employed to quantify early and
late
apoptotic cells. The results demonstrated that treatment with AKBA at various concentrations (2.5, 5.0,
and 10.0
µM) for 24 hours significantly increased the number of apoptotic cells in mature 3T3-L1 adipocytes
compared to
the control group (P<0.01, Fig. 4B). To further investigate the molecular event by which AKBA affects
apoptosis, we evaluated the expression of pro-and anti-apoptotic proteins Bax and Bcl-2 which are critical
regulators of the mitochondrial apoptotic pathway [35] by western blotting (Fig. 4C). AKBA treatment
tended to
increase Bax expression, while 10 μM AKBA markedly reduced Bcl-2 levels (P<0.01, Fig. 4D) resulting in
an
elevated Bax/Bcl2 ratio (P<0.05, Fig. 4D) and enhanced apoptosis. The increased apoptosis following
AKBA
treatment may account for the observed decrease in cell viability and lipid accumulation in this study.
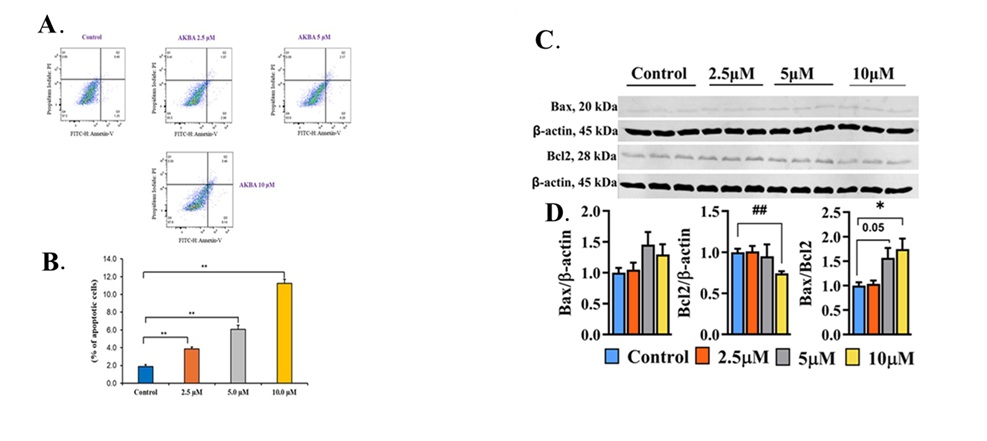
Fig. 4: AKBA treatment promoted apoptosis in 3T3-L1 mature adipocytes. (A) Annexin-PI staining in 3T3-L1cells after treatment with AKBA. (B) The percentage of apoptotic cells in 3T3-L1 cells after treatment with AKBA. Q1: (AnnexinV− FITC) ⁻/PI⁺, necrotic cells or a few late apoptotic cells, mechanically damaged cells; Q2: (AnnexinV+ FITC) ⁺/PI⁺, late apoptotic cells; Q3: (AnnexinV− FITC) ⁺/PI⁻, early late apoptotic cells; Q4: (AnnexinV− FITC) ⁻/PI⁻, live cell. Data are reported as mean ±SD, n=3. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test. **P<0.01vs. control group. (C) Immunoblots for Bax and Bcl2 with β-actin as loading control. (D) The bar graph represents protein band density. Data are represented as Mean ± SEM, n=3, and reported as fold change compared to the control. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test; * P < 0.05, and unpaired Student’s t-test; ## P < 0.01. P values < 0.05 were considered statistically significant for all comparisons.
AKBA reduced adipogenic and lipogenic factors in 3T3-L1 cells
PPARγ and C/EBPα are essentially involved in adipocyte differentiation [8]. They can induce
adipose-specific
genes, including fatty acid synthase (FAS), which is responsible for lipid synthesis [9]. We evaluated the
expression levels of C/EBPβ, PPARγ, and C/EBPα, key transcription factors implicated in adipocyte
differentiation, to understand the mechanism of AKBA-mediated reduction in intracellular lipid content.
2.5 µM,
5 µM, and 10 µM of AKBA treatment significantly reduced the protein levels of C/EBPβ (10%-55%), PPARγ (20%
to
65%), and C/EBPα (20%-70%) and lipogenic factor FAS (18 to 55%) compared to control treatment (Fig. 5A and
5B).
These findings suggest that AKBA-mediated reduction of adipogenic and lipogenic transcription factors may
be
accountable in part for the lowered lipid content detected after AKBA administration.
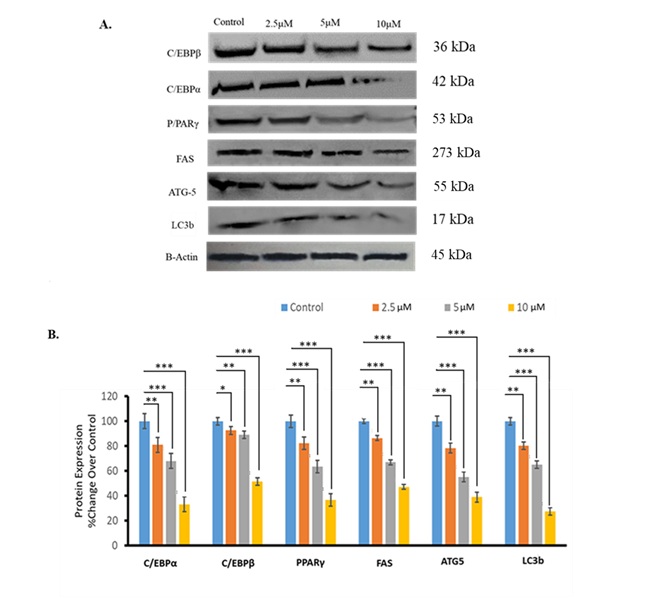
Fig. 5: AKBA treatment affected expression of adipocyte differentiation, autophagy, and lipogenic markers. (A) Immunoblots for proteins related to adipocyte differentiation, autophagy, and lipid accumulation. Representative blots are shown. (B) The bar graph represents protein band density. Data are mean ± SD and reported as percentage change compared to the control. n=3. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test. *P<0.5, **P<0.01, ***P<0.001 vs. control. P values < 0.05 were considered statistically significant for all comparisons.
AKBA suppressed protein expression linked to autophagy in 3T3-L1 adipocytes
Autophagy has a substantial influence on adipocyte development [36]. Thus, we checked the expression of
autophagy-related proteins following AKBA treatment. AKBA treatment with 2.5 µM, 5 µM, and 10 µM markedly
reduced ATG5 protein expression by 20%, 40%, and 50%, respectively, compared to the control treatment
(P<0.01, P<0.001, P<0.001, respectively, Fig. 5A and 5B). The same AKBA treatment reduced LC3b
protein
expression by 20%, 35%, and 65%, respectively than the control treatment (P<0.01, P<0.001,
P<0.001,
respectively, Fig. 5A and 5B). These results indicate that AKBA has the potential to suppress autophagy in
adipocytes.
AKBA increased AMPK phosphorylation in 3T3-L1 cells
AMP-activated protein kinase (AMPK) is crucial for adipocyte proliferation, differentiation, and lipid
metabolism [37]. Additionally, increased AMPK phosphorylation, an indicator of AMPK activation, has
demonstrated
anti-adipogenic effects [37]. Next, we evaluated whether AKBA-mediated repression of adipogenic
transcription
factor expression and lipid accumulation in mature adipocytes involves AMPK activation. Western Blot
results
indicated that the phosphorylation of AMPK was significantly enhanced 30%, 60%, and 100% upon treatment
with 2.5
µM, 5 µM, and 10 µM of AKBA (P<0.01, P<0.01, and P<0.001, respectively, Fig. 6A and 6B). The
findings
suggest that AKBA promotes AMPK phosphorylation (Thr172), which may contribute to blocking adipocyte
differentiation and reducing lipid accumulation in 3T3-L1 cells.
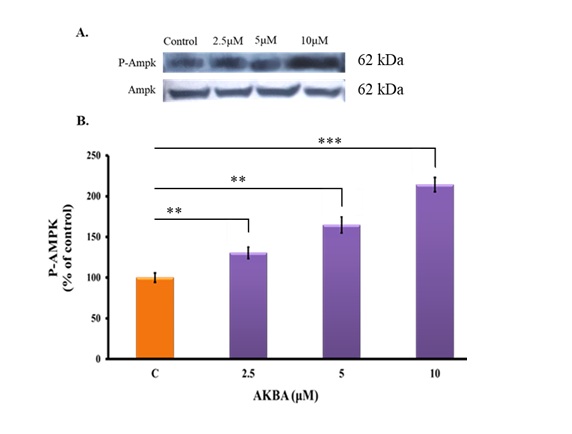
Fig. 6: Effects of AKBA on AMPK phosphorylation in 3T3-L1 cells. (A) Immunoblot for P-AMPK. Representative blots are shown. (B) The bar graph represents protein band density. Data are mean ±SD and presented as percentage change over control following normalization to AMPK. n=3. Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc test. **P<0.01, **P<0.001 vs. control group. P values < 0.05 were considered statistically significant for all comparisons.
Molecular docking results
The molecular docking was performed to explore the atomic interactions between the PPARγ (PDB ID:
6C1I)
and ATG5
(PDB ID: 7W36) proteins retrieved from the protein databank server (Fig. 7A). The molecular interactions
results
reveal critical interactions between the reference ligand 6C1I and PPARγ (T0070907), with noteworthy
bonds
at
SER289 and HIS323, both involving hydrogen bonds that stabilize the ligand within the binding site (Fig.
7B).
For the ATG5 peptide inhibitor 7W36, significant ionic bonds are observed with Asp3 from both Lys3 and
Arg4 and
a powerful bond with Asp10 from Arg11, indicating key sites for drug targeting (Fig. 7C). Additionally,
hydrogen
bond interactions involving Arg13, Gln17, His5, and Trp2 of the stapled peptide with ATG5 active site
residues
suggest a strong binding profile that could be pivotal for inhibitory efficacy (Fig. 7D). The
co-crystalized ligand of the PPARγ was redocked in the active pocket of the protein to validate the
docking
protocol and shows RMSD value of 0.684 Å with a docking score of -5.67 kcal/mol, which shows a favorable
RMSD
and accuracy of the docking protocol (Fig. 8A). The AKBA, our compound of interest, demonstrates potent
interactions with PPARγ, achieving a docking score of -7.67 kcal/mol, and with ATG5, where it
attains a
docking
score of -6.57 kcal/mol. AKBA demonstrates potent interactions with both PPARγ and ATG5 active site
residues. In
the PPARγ active site, AKBA forms hydrogen bonds with TYR327 and GLN286, positioning it effectively
within
the
binding domain (Fig. 8B). With ATG5, AKBA shows strong and repeated interactions with ARG41, noticeable
both
hydrogen- and ionic bonds, suggesting a highly favorable binding affinity and specificity (Fig. 8C). These
interactions, particularly those with high binding energies, are crucial for understanding the molecular
basis
of AKBA’s action and its potential as a targeting therapeutic agent against PPARγ and ATG5. The
detailed
interactions of the reference inhibitors and AKBA with PPARγ and ATG5 are shown in Table 1.
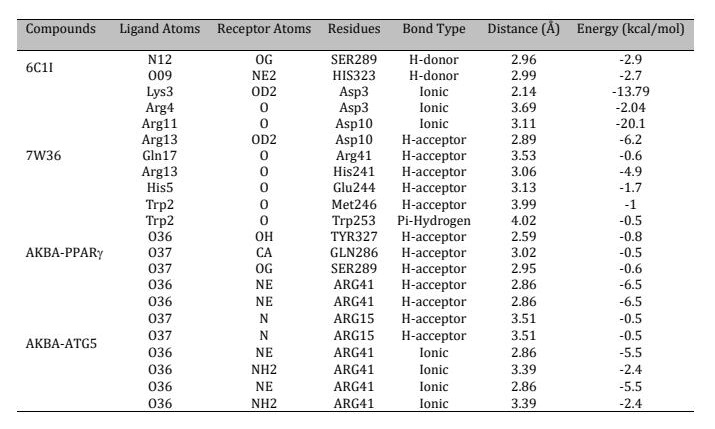
Table 1: Summary of molecular docking interactions of the reference ligand 6C1I with PPAR, the ATG5 peptide inhibitor with active site residues, and the interactions of AKBA with the active site residues of both PPAR and ATG5
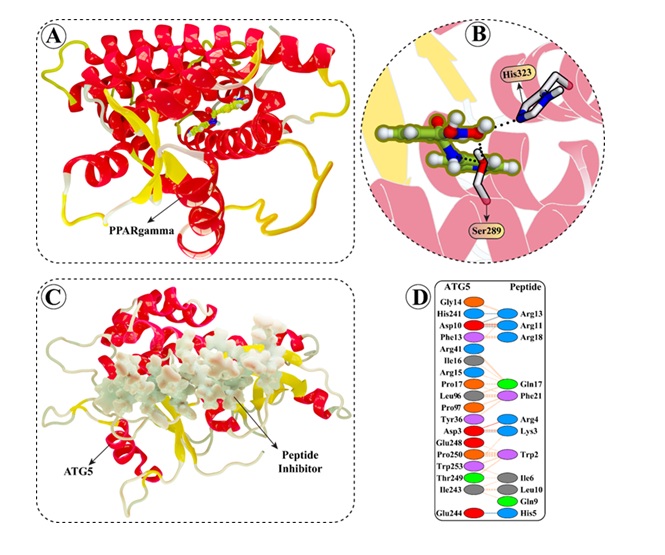
Fig. 7: Detailed visual representation of the (A) PPARg protein having (B) T0070907 ligand attached in the active site and (C) ATG5 structures having (D) stapled peptide in the active site retrieved from the protein data bank server with PDB IDs 6C1I and 7W36.
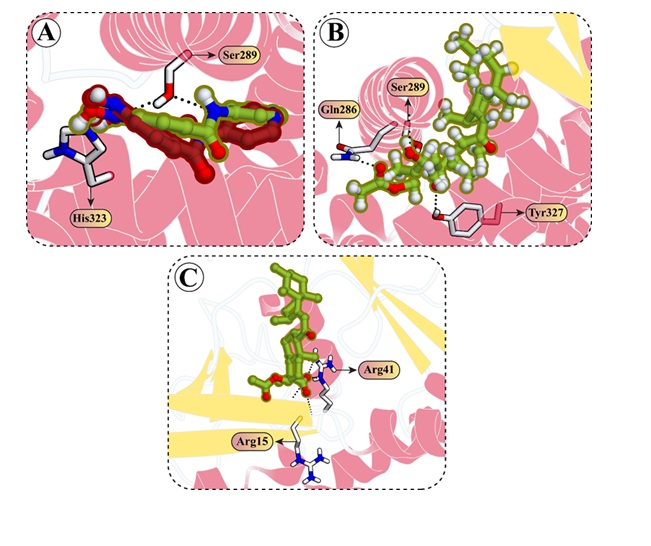
Fig. 8: Docking interactions of AKBA with PPARg and ATG5 proteins. (A) The co-crystallized ligand of PPARg, having PDB ID 6C1I, was redocked, resulting in an RMSD value of 0.684 Å between the co-crystallized (shown in green) and redocked (shown in red) structures. (B) 3D visualization of the interaction between AKBA and the active site residues of PPARg. (C) Molecular interactions of AKBA with the active site residues of the ATG5 protein.
Discussion
The current study uncovered that AKBA blocked adipocyte development by lowering the expression of adipogenic gene regulators C/EBPα, C/EBPβ, and PPARγ. Furthermore, AKBA inhibited lipid formation in 3T3-L1 cells while downregulating lipogenic protein FAS expression.
Crucially, AKBA treatment stimulated AMPK phosphorylation and repressed autophagy-related proteins ATG5 and LC3b. Molecular docking studies showed that AKBA significantly docked with PPARγ and ATG5. Our study is the first to demonstrate the involvement of autophagy and AMPK in controlling adipocyte differentiation.
Obesity is a public health issue that arises from the enlargement (hypertrophy) and increase in number (hyperplasia) of adipocytes, as well as immune cell infiltration in adipose tissue [38]. Significantly, apoptosis in both progenitor and mature adipocyte cells plays a crucial role in regulating overall body adipose mass [39]. Several natural compounds have shown promise in inducing apoptosis in 3T3-L1 adipocytes [40-42]. Mechanistically, apoptosis can be triggered through two distinct signaling pathways: the extrinsic pathway, which involves death receptors on the cell surface, and the intrinsic pathway, which operates through the mitochondria [43]. Bax and Bcl2 are key mitochondrial apoptosis regulators, with Bcl-2 functioning as an anti-apoptotic protein and Bax as a pro-apoptotic mediator. The elevated Bax/Bcl2 ratio is a critical determinant of apoptosis induction [44]. In the present study, AKBA treatment demonstrated an increased trend of Bax and decreased Bcl2 expression, elevating the Bax/Bcl2 ratio in mature 3T3-L1 adipocytes, suggesting that AKBA-induced apoptosis involves Bcl2 family proteins and the mitochondrial pathway. Several bioactive compounds were found effective in inducing apoptosis in mature adipocytes by regulating Bax and Bcl2 [42, 45]. However, the contribution of cytochrome C and caspase-3, both central regulators of apoptosis, remains undefined in AKBA-mediated apoptosis and warrants further investigation. Further, AMPK activation can trigger proapoptotic protein expression by regulating AKT [46]. We observed that AKBA increases AMPK phosphorylation and induces apoptosis in mature adipocytes, potentially contributing to reduced cell viability and inhibited lipid accumulation. However, the precise mechanisms by which AKBA triggers apoptosis in mature adipocytes remain unclear and require further investigation.
Transcription factors and signaling pathways critically regulate adipogenesis. In early differentiation, expression levels of C/EBPβ/δ are high, stimulating PPARγ and C/EBPα, essential transcription factors for adipogenesis [8]. During differentiation's terminal stage, PPARγ and C/EBPα induce adipocyte-specific genes such as FAS and LPL [7]. FAS is crucial for fatty acid production in 3T3-L1 adipocytes [47]. AKBA treatment markedly reduced these factors at the protein level. We have demonstrated that AKBA dose-dependently reduced the expression of C/EBPβ, PPARγ, and C/EBPα and the lipogenic protein FAS, which may be responsible for AKBA-mediated inhibition of adipocyte differentiation and lipid accumulation in differentiated adipocytes. Liu et al. previously reported AKBA-mediated reduction of adipogenic transcription factors and induction of lipolysis in 3T3-L1 cells [48]. In their study, AKBA was applied to fully differentiated adipocytes, whereas in ours, 3T3-L1 preadipocytes were treated with AKBA during the differentiation process. Our findings show that AKBA inhibits adipogenesis by suppressing C/EBPβ, C/EBPα, and PPAR-γ, while concurrently inducing apoptosis.
Autophagy, a critical cellular breakdown system, begins with the production of autophagosomes, which then destroy organelles and proteins [49]. Autophagy activation and inhibition are associated with a large number of pathophysiological conditions, such as cancer, diabetes, and especially neurodegenerative disorders [50]. New evidence indicates that autophagy has an essential part in lipid buildup in fat cells [51]. Singh et al. found that inhibiting autophagy limited TG accumulation in 3T3-L1 adipocytes and suppressed the expression of adipocyte differentiation marker proteins [52]. Notably, autophagy inactivation via ATG7 deletion altered adipogenesis, leading to the development of adipose tissue with a variety of anti-obesity and anti-diabetic features. This study not only demonstrated autophagy's physiological importance in adipogenesis, but it may also open up intriguing possibilities for treating obesity and diabetes [53, 54].
A study using primary MEFs revealed that autophagy is induced during adipogenesis, and atg5 deficiency drastically lowered adipogenesis efficiency. While ATG5 deletion did not significantly affect early events, it may cause adipogenesis arrest at later stages and can lead to apoptosis. The study also demonstrated that ATG5 function is crucial for adipogenesis, suggesting autophagy's involvement in adipogenesis [53, 54]. Interestingly, one study demonstrated that autophagy genes are controlled during the development of adipocytes [55]. Notably, this regulation was mediated by adipogenic transcription factors like C/EBPβ and PPARγ. The AKBA-mediated reduction in ATG5 observed in our study could explain the lower expression of the adipocyte differentiation marker, suppression of adipocyte differentiation, and lower lipid levels in adipocytes.
AMPK is a metabolic sensor that governs cellular energy homeostasis [56]. Its activation inhibits fatty acids, cholesterol, and triglycerides synthesis and stimulates fatty acid oxidation. Additionally, AMPK inhibits ACC1, SREBP1c, and FAS, preventing the accumulation of lipids and sterols [57]. Significantly, AMPK activation can inhibit adipocyte differentiation by repressing the crucial adipogenic transcription factors, including C/EBPα and PPARγ [58]. Our study revealed that AKBA activated AMPK phosphorylation while suppressing autophagy proteins ATG5 and LC3b and inhibited adipocyte differentiation. Important evidence suggests that AMPK activation also stimulates autophagy activity [59]. Discrepancies in findings regarding the impact of AMPK activation on autophagy and adipogenesis exist. Rahman et al. showed that Bifidobacterium longum subsp. infantis YB0411 (YB) treatment inhibited adipocyte differentiation by suppressing adipogenic transcription factors such as C/EBPα, C/EBPβ, and PPARγ. Further, YB-mediated blockade of adipocyte differentiation was concomitant with the suppression of autophagy marker p62 and LC3B and AMPK activation [60]. In contrast, Li et al. found that icariin, a flavonoid, blocked adipocyte differentiation by suppressing AMPK activation and autophagy [61].
Overall, our findings suggest that AKBA may inhibit adipogenesis through AMPK activation and autophagy regulation and via apoptosis induction.
Limitations of our study
Despite the demonstrated effectiveness of AKBA in inhibiting adipocyte differentiation and lipid
accumulation,
this study has several limitations. First, the precise roles of AMPK and autophagy in AKBA-mediated
regulation
of adipogenesis remain unclear. Although AKBA suppressed autophagy markers in adipocytes, its direct
impact on
adipogenesis remains unclear, partly because autophagy is a dynamic process and static measurements of
ATG5 and
LC3b do not adequately reflect autophagic flux. Therefore, future studies should employ lysosomal
inhibitors
such as bafilomycin A1 or chloroquine to more reliably assess autophagy and clarify the role of AKBA in
regulating adipocyte differentiation. Additionally, comparing AKBA’s binding affinities with known PPARγ
and
ATG5 ligands, along with conducting molecular dynamics simulations, would strengthen the biological
relevance of
our docking results. These aspects should be addressed in future studies. Finally, while the present study
was
limited to in vitro experiments, in vivo outcomes may differ due to factors such as
pharmacokinetics and tissue distribution. Therefore, future animal studies are planned to evaluate the
anti-obesity effects of AKBA, with particular emphasis on its bioavailability and impact on adipose
tissue.
Conclusion
AKBA inhibited adipocyte differentiation by repressing adipogenic factors such as C/EBPα, C/EBPβ, and PPARγ. Further, AKBA induced apoptosis by increasing the Bax/Bcl2 and reducing lipid accumulation and FAS protein expression in differentiated adipocytes. Importantly, AKBA induced AMPK phosphorylation and suppressed autophagy-related proteins ATG5 and LC3b. Molecular docking studies have demonstrated the proficient binding of AKBA to PPARγ and ATG5 (Fig. 9). Altogether, the efficacy of AKBA in inhibiting adipogenesis has been demonstrated through its targeting of AMPK, PPARγ, ATG5, and the mitochondrial apoptotic pathway. These findings underscore the potential of AKBA as a natural anti-obesity therapeutic agent. Nevertheless, further in vivo research is necessary to validate these in vitro findings.
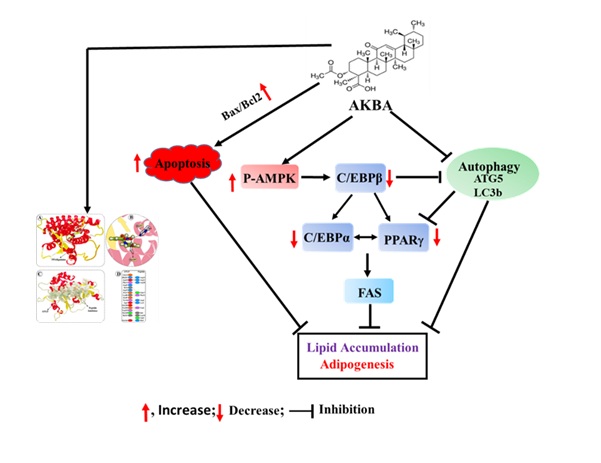
Fig. 9: An illustration of how AKBA regulates adipogenesis. AKBA treatment may inhibit adipocyte differentiation factors (C/EBPβ and PPARg) and lipid accumulation by inducing AMPK phosphorylation and suppressing autophagy in 3T3-L1 cells. Further, AKBA may promote apoptosis by increasing the Bax/Bcl2 ratio in 3T3-L1 cells.
Abbreviations
AKBA: 3-acetyl-11-keto-β-Boswellic acid; ATG5: autophagy related 5 gene; C/EBPs: CCAAT/enhancer-binding proteins; LC3b: microtubule-associated protein 1 light chain 3b; PPARγ: peroxisome proliferator-activated receptor gamma; AMPK: AMP-activated protein kinase; HbA1c: Hemoglobin A1c; FAS: fatty-acid-synthase; LPL: lipoprotein lipase
Acknowledgements
The authors thank Dr. Nitta Shree (Texas A&M University, USA) for her assistance in generating the immunoblot data for Bcl2 and Bax proteins. We also thank Ms. Lauren Gladwell (Texas A&M University, USA) for her help in editing the manuscript.
Funding
This study was supported by a grant from The Oman Research Council (TRC)(Grant # BFP/RGP/HSS/24/201).
Author Contributions
Faizullah Khan: Conceptualization, Data curation, Statistical analysis, Writing- Original
draft
preparation, read and approved the final manuscript. Muhammad Waqas: Methodology, Data
curation, read and approved the final manuscript. Hassan Moghtaderi: Methodology, read
and
approved the final manuscript. Najeeb Ur Rehman: Methodology, Writing - review & editing,
read
and approved the final manuscript. Satya Kumar Avula: Methodology, Writing - review &
editing,
read and approved the final manuscript. Haroon Khan: Writing - review & editing, read and
approved
the final manuscript. Mahua Choudhury: Writing - review & editing, read and approved the
final
manuscript. Ahmed Al-Harrasi: Writing - review & editing, read and approved the final
manuscript. Shaikh Mizanoor Rahman: Conceptualization, Supervision, Writing- Original draft
preparation, Writing-review & editing, read and approved the final manuscript. All authors read
and
approved the final version of the manuscript.
Data availability
Data will be made available on request.
AI Disclosure
No AI tools have been used to create this work.
Disclosure Statement
The authors declare that they have no conflicts of interest.
References
| 1 | Hossain, P., B. Kawar, and M. El Nahas, Obesity and diabetes in the developing world-a growing
challenge. New England journal of medicine, 2007.356(3): p. 213-215.
https://doi.org/10.1056/NEJMp068177 |
| 2 | Lam, Y.Y. and E. Ravussin, Analysis of energy metabolism in humans: A review of methodologies.
Molecular metabolism, 2016.5(11): p. 1057-1071.
https://doi.org/10.1016/j.molmet.2016.09.005 |
| 3 | Mokdad, A.H., et al., The continuing epidemics of obesity and diabetes in the United States. Jama,
2001.286(10): p. 1195-1200.
https://doi.org/10.1001/jama.286.10.1195 |
| 4 | Hubert, H.B., et al., Obesity as an independent risk factor for cardiovascular disease: a 26-year
follow-up of participants in the Framingham Heart Study. Circulation, 1983.67(5): p. 968-977.
https://doi.org/10.1161/01.CIR.67.5.968 |
| 5 | Smith, U. and B.B. Kahn, Adipose tissue regulates insulin sensitivity: role of adipogenesis, de
novo lipogenesis and novel lipids. Journal of internal medicine, 2016.280(5): p. 465-475.
https://doi.org/10.1111/joim.12540 |
| 6 | Farmer, S.R., Transcriptional control of adipocyte formation. Cell metabolism, 2006.4(4): p.
263-273.
https://doi.org/10.1016/j.cmet.2006.07.001 |
| 7 | Rosen, E.D., et al., Transcriptional regulation of adipogenesis. Genes & development, 2000.14(11):
p. 1293-1307.
https://doi.org/10.1101/gad.14.11.1293 |
| 8 | Guo, L., X. Li, and Q.-Q. Tang, Transcriptional regulation of adipocyte differentiation: a central
role for CCAAT/enhancer-binding protein (C/EBP) β. Journal of biological chemistry, 2015.290(2): p.
755-761.
https://doi.org/10.1074/jbc.R114.619957 |
| 9 | de Sá, P.M., et al., Transcriptional regulation of adipogenesis. Comprehensive Physiology,
2011.7(2): p. 635-674.
https://doi.org/10.1002/j.2040-4603.2017.tb00753.x |
| 10 | Abdul, W.M., et al., Therapeutic role of Ricinus communis L. and its bioactive compounds in
disease prevention and treatment. Asian pacific journal of tropical medicine, 2018.11(3): p.
177-185.
https://doi.org/10.4103/1995-7645.228431 |
| 11 | Khanna, D., et al., Natural products as a gold mine for arthritis treatment. Current opinion in
pharmacology, 2007.7(3): p. 344-351.
https://doi.org/10.1016/j.coph.2007.03.002 |
| 12 | Liu, J.-J., et al., Keto-and acetyl-keto-boswellic acids inhibit proliferation and induce
apoptosis in Hep G2 cells via a caspase-8 dependent pathway. International journal of molecular
medicine, 2002.10(4): p. 501-505.
https://doi.org/10.3892/ijmm.10.4.501 |
| 13 | Liu, J.-J., et al., Boswellic acids trigger apoptosis via a pathway dependent on caspase-8
activation but independent on Fas/Fas ligand interaction in colon cancer HT-29 cells.
Carcinogenesis, 2002.23(12): p. 2087-2093.
https://doi.org/10.1093/carcin/23.12.2087 |
| 14 | Sengupta, K., et al., A double blind, randomized, placebo controlled study of the efficacy and
safety of 5-Loxin® for treatment of osteoarthritis of the knee. Arthritis research & therapy,
2008.10(4): p. 1-11.
https://doi.org/10.1186/ar2461 |
| 15 | Al-Harrasi, A., et al., Chemistry and bioactivity of boswellic acids and other terpenoids of the
genus boswellia. 2018: Elsevier.
https://doi.org/10.1016/B978-0-08-102441-6.00002-5 |
| 16 | Shamraiz, U., et al., Synthesis of new boswellic acid derivatives as potential antiproliferative
agents. Natural product research, 2020.34(13): p. 1845-1852.
https://doi.org/10.1080/14786419.2018.1564295 |
| 17 | Shehata, A.M., et al., Prevention of multiple low-dose streptozotocin (MLD-STZ) diabetes in mice
by an extract from gum resin of Boswellia serrata (BE). Phytomedicine, 2011.18(12): p. 1037-1044.
https://doi.org/10.1016/j.phymed.2011.06.035 |
| 18 | Shehata, A., et al., 11-keto-β-boswellic acids prevent development of autoimmune reactions,
insulitis and reduce hyperglycemia during induction of multiple low-dose streptozotocin (MLD-STZ)
diabetes in mice. Hormone and Metabolic Research, 2015.47(06): p. 463-469.
https://doi.org/10.1055/s-0035-1547293 |
| 19 | Gomaa, A.A., et al., Inhibition of adiposity and related metabolic disturbances by polyphenol-rich
extract of Boswellia serrata gum through alteration of adipo/cytokine profiles.
Inflammopharmacology, 2019.27: p. 549-559.
https://doi.org/10.1007/s10787-018-0519-4 |
| 20 | Ahangarpour, A., et al., Effect of Boswellia serrata supplementation on blood lipid, hepatic
enzymes and fructosamine levels in type2 diabetic patients. Journal of Diabetes & Metabolic
Disorders, 2014.13: p. 1-5.
https://doi.org/10.1186/2251-6581-13-29 |
| 21 | Azadmehr, A., et al., A randomized clinical trial study: anti-oxidant, anti-hyperglycemic and
anti-hyperlipidemic effects of olibanum gum in type 2 diabetic patients. Iranian journal of
pharmaceutical research: IJPR, 2014.13(3): p. 1003.
|
| 22 | Rehman, N.U., et al., New α-glucosidase inhibitors from the resins of Boswellia species with
structure-glucosidase activity and molecular docking studies. Bioorganic chemistry, 2018.79: p.
27-33.
https://doi.org/10.1016/j.bioorg.2018.04.020 |
| 23 | Khan, A., et al., Loading AKBA on surface of silver nanoparticles to improve their
sedative-hypnotic and anti-inflammatory efficacies. Nanomedicine, 2019.14(21): p. 2783-2798.
https://doi.org/10.2217/nnm-2019-0211 |
| 24 | Rahman, S.M., et al., CCAAT/enhancer-binding protein β (C/EBPβ) expression regulates
dietary-induced inflammation in macrophages and adipose tissue in mice. Journal of Biological
Chemistry, 2012.287(41): p. 34349-34360.
https://doi.org/10.1074/jbc.M112.410613 |
| 25 | Rahman, S.M., et al., Fenofibrate and PBA prevent fatty acid-induced loss of adiponectin receptor
and pAMPK in human hepatoma cells and in hepatitis C virus-induced steatosis. Journal of lipid
research, 2009.50(11): p. 2193-2202.
https://doi.org/10.1194/jlr.M800633-JLR200 |
| 26 | ULC, C.C.G., Molecular Operating Environment (MOE). 2022, Chemical Computing Group ULC: 1010
Sherbooke St. West, Suite 910, Montreal, QC, Canada.
|
| 27 | Gerber, P.R. and K. Muller, MAB, a generally applicable molecular force field for structure
modelling in medicinal chemistry. J Comput Aided Mol Des, 1995.9(3): p. 251-68.
https://doi.org/10.1007/BF00124456 |
| 28 | Maier, J.A., et al., ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters
from ff99SB. J Chem Theory Comput, 2015.11(8): p. 3696-713.
https://doi.org/10.1021/acs.jctc.5b00255 |
| 29 | Kanehisa, M., et al., From genomics to chemical genomics: new developments in KEGG. Nucleic acids
research, 2006.34(suppl_1): p. D354-D357.
https://doi.org/10.1093/nar/gkj102 |
| 30 | Drews, J., Drug discovery: a historical perspective. science, 2000 287(5460): p. 1960-1964.
https://doi.org/10.1126/science.287.5460.1960 |
| 31 | Chyau, C.-C., et al., The inhibitory effects of djulis (Chenopodium formosanum) and its bioactive
compounds on adipogenesis in 3T3-L1 adipocytes. Molecules, 2018.23(7): p. 1780.
https://doi.org/10.3390/molecules23071780 |
| 32 | Hong, J.W., et al., Inhibition of Lipase Activity and Preadipocyte Differentiation in3T3-L1 Cells
Treated with Sargassum horneri Extract. Ocean and Polar Research, 2022.44(1): p. 61-67.
|
| 33 | Blanco, J., et al., Obesogenic effects of chlorpyrifos and its metabolites during the
differentiation of 3T3-L1 preadipocytes. Food and Chemical Toxicology, 2020.137: p. 111171.
https://doi.org/10.1016/j.fct.2020.111171 |
| 34 | Vermes, I., et al., A novel assay for apoptosis flow cytometric detection of phosphatidylserine
expression on early apoptotic cells using fluorescein labelled annexin V. Journal of immunological
methods, 1995.184(1): p. 39-51.
https://doi.org/10.1016/0022-1759(95)00072-I |
| 35 | Gross, A., BCL‐2 proteins: Regulators of the mitochondrial apoptotic program. IUBMB life,
2001.52(3‐5): p. 231-236.
https://doi.org/10.1080/15216540152846046 |
| 36 | Khan, F., et al., Autophagy in adipogenesis: Molecular mechanisms and regulation by bioactive
compounds. Biomedicine & Pharmacotherapy, 2022. 155: p. 113715.
https://doi.org/10.1016/j.biopha.2022.113715 |
| 37 | Ceddia, R., The role of AMP-activated protein kinase in regulating white adipose tissue
metabolism. Molecular and Cellular Endocrinology, 2013.366(2): p. 194-203.
https://doi.org/10.1016/j.mce.2012.06.014 |
| 38 | Crewe, C., Y.A. An, and P.E. Scherer, The ominous triad of adipose tissue dysfunction:
inflammation, fibrosis, and impaired angiogenesis. The Journal of clinical investigation,
2017.127(1): p. 74-82.
https://doi.org/10.1172/JCI88883 |
| 39 | Sorisky, A., R. Magun, and A. Gagnon, Adipose cell apoptosis: death in the energy depot.
International Journal of Obesity, 2000.24(4): p. S3-S7.
https://doi.org/10.1038/sj.ijo.0801491 |
| 40 | Choi, E.J., J.Y. Jung, and G.-H. Kim, Genistein inhibits the proliferation and differentiation of
MCF-7 and 3T3-L1 cells via the regulation of ERα expression and induction of apoptosis. Experimental
and therapeutic medicine, 2014 8(2): p. 454-458.
https://doi.org/10.3892/etm.2014.1771 |
| 41 | Yang, J.Y., et al., Molecular mechanisms of apoptosis induced by ajoene in 3T3‐L1 adipocytes.
Obesity, 2006.14(3): p. 388-397.
https://doi.org/10.1038/oby.2006.52 |
| 42 | Yang, J.-Y., M.A. Della-Fera, and C.A. Baile, Esculetin induces mitochondria-mediated apoptosis in
3T3-L1 adipocytes. Apoptosis, 2006.11(8): p. 1371-1378.
https://doi.org/10.1007/s10495-006-7691-5 |
| 43 | Newton, K., et al., Cell death. Cell, 2024.187(2): p. 235-256.
https://doi.org/10.1016/j.cell.2023.11.044 |
| 44 | Siddiqui, W.A., A. Ahad, and H. Ahsan, The mystery of BCL2 family: Bcl-2 proteins and apoptosis:
an update. Archives of toxicology, 2015.89(3): p. 289-317.
https://doi.org/10.1007/s00204-014-1448-7 |
| 45 | Zhu, L., et al., Curcumin triggers apoptosis via upregulation of Bax/Bcl-2 ratio and caspase
activation in SW872 human adipocytes. Molecular medicine reports, 2015.12(1): p. 1151-1156.
https://doi.org/10.3892/mmr.2015.3450 |
| 46 | Kim, H.-J., et al., Red pepper seed water extract inhibits preadipocyte differentiation and
induces mature adipocyte apoptosis in 3T3-L1 cells. Nutrition research and practice, 2018.12(6): p.
494-502.
https://doi.org/10.4162/nrp.2018.12.6.494 |
| 47 | Ronnett, G.V., et al., Fatty acid metabolism as a target for obesity treatment. Physiology &
behavior, 2005.85(1): p. 25-35.
https://doi.org/10.1016/j.physbeh.2005.04.014 |
| 48 | Liu, J.-J., et al., Acetyl-keto-β-boswellic acid induces lipolysis in mature adipocytes.
Biochemical and Biophysical Research Communications, 2013.431(2): p. 192-196.
https://doi.org/10.1016/j.bbrc.2012.12.136 |
| 49 | Klionsky, D.J., Autophagy: from phenomenology to molecular understanding in less than a decade.
Nature reviews Molecular cell biology, 2007.8(11): p. 931-937.
https://doi.org/10.1038/nrm2245 |
| 50 | Ichimiya, T., et al., Autophagy and autophagy-related diseases: a review. International journal of
molecular sciences, 2020.21(23): p. 8974.
https://doi.org/10.3390/ijms21238974 |
| 51 | Levine, B. and J. Yuan, Autophagy in cell death: an innocent convict? The Journal of clinical
investigation, 2005 115(10): p. 2679-2688.
https://doi.org/10.1172/JCI26390 |
| 52 | Singh, R., et al., Autophagy regulates adipose mass and differentiation in mice. The Journal of
clinical investigation, 2009.119(11): p. 3329-3339.
https://doi.org/10.1172/JCI39228 |
| 53 | Zhang, Y., et al., Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a
role in adipogenesis. Proceedings of the National Academy of Sciences, 2009.106(47): p. 19860-19865.
https://doi.org/10.1073/pnas.0906048106 |
| 54 | Baerga, R., et al., Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a
cellular model and in mice. Autophagy, 2009.5(8): p. 1118-1130.
https://doi.org/10.4161/auto.5.8.9991 |
| 55 | Ahmed, M., et al., Transcriptional regulation of autophagy genes via stage-specific activation of
CEBPB and PPARG during adipogenesis: a systematic study using public gene expression and
transcription factor binding datasets. Cells, 2019.8(11): p. 1321.
https://doi.org/10.3390/cells8111321 |
| 56 | Carling, D., The AMP-activated protein kinase cascade-a unifying system for energy control. Trends
in biochemical sciences, 2004.29(1): p. 18-24.
https://doi.org/10.1016/j.tibs.2003.11.005 |
| 57 | Hardie, D.G., F.A. Ross, and S.A. Hawley, AMPK: a nutrient and energy sensor that maintains energy
homeostasis. Nature reviews Molecular cell biology, 2012.13(4): p. 251-262.
https://doi.org/10.1038/nrm3311 |
| 58 | Fernández‐Veledo, S., et al., Role of energy‐and nutrient‐sensing kinases AMP‐activated Protein
Kinase (AMPK) and Mammalian Target of Rapamycin (mTOR) in Adipocyte Differentiation. IUBMB life,
2013.65(7): p. 572-583.
https://doi.org/10.1002/iub.1170 |
| 59 | Li, Y. and Y. Chen, AMPK and autophagy. Autophagy: Biology and Diseases: Basic Science, 2019: p.
85-108.
https://doi.org/10.1007/978-981-15-0602-4_4 |
| 60 | Rahman, M.S., et al., Bifidobacterium longum subsp. infantis YB0411 inhibits adipogenesis in
3T3-L1 pre-adipocytes and reduces high-fat-diet-induced obesity in mice. Journal of Agricultural and
Food Chemistry, 2021.69(21): p. 6032-6042.
https://doi.org/10.1021/acs.jafc.1c01440 |
| 61 | Li, H., et al., Icariin inhibits AMPK-dependent autophagy and adipogenesis in adipocytes in vitro
and in a model of graves' orbitopathy in vivo. Frontiers in physiology, 2017.8: p. 45.
https://doi.org/10.3389/fphys.2017.00045 |