×
![]()
Corresponding Author: Namir Shaabani and John R. Teijaro
Department of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, CA, USA
E-Mail shaabani@scripps.edu; teijaro@scripps.edu
IFN-β, but not IFN-α, is Responsible for the Pro-Bacterial Effect of Type I Interferon
Namir Shaabania Vincent F. Vartabediana Nhan Nguyena Nadine Honkeb Zhe Huanga John R. Teijaroa
aDepartment of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, CA, USA, bDepartment of Rheumatology, Hiller Research Center Rheumatology, University Hospital Düsseldorf, Düsseldorf, Germany
Introduction
Although originally discovered and characterized as a potent antiviral protein, recent studies have shown that type I Interferon (IFN-I) signaling also induces detrimental effects during infection. During acute viral infection, such effects include morbidity and mortality through recruitment of inflammatory innate immune cell populations and induction of aberrant inflammatory responses [1, 2]. During chronic viral infection, IFN-I signaling initiates and maintains immune suppression, lymphoid tissue disorganization, and T cell dysfunction through modulation of multiple immune cell populations [3-5]. Similarly, during bacterial infection, IFN-I signaling has been reported to both contribute to protection and increase susceptibility to infection [6-8]. Additionally, elevated IFN-I production following viral infection has been causally linked with promoting host susceptibility to secondary bacterial infections [9-14] which can result in severe morbidity and mortality.
The IFN-I family is composed of several IFN-I subtypes (13 in humans and 14 in mice) including multiple IFN-α homologues, IFN-β, and lesser-studied subtypes like IFN-ε, IFN-ω, and IFN-қ [15]. A sequencing comparison between IFN-α and IFN-β showed nearly 50% amino acid homology, while IFN-α subtypes are about 80% homologous to each other [16]. In addition to differential sequences, each IFN-I subtype has different signaling signatures, biological properties, and therapeutic utility. For instance, IFN-α2 is used clinically to treat and cure viral hepatitis [17] while IFN-β is used to reduce pathology associated with multiple sclerosis [18].
Despite these differences, all IFN-I subtypes share the same multicomponent cell surface receptor, the interferon-α/β receptor (IFNAR), with IFN-β having the highest binding affinity to IFNAR [19]. IFNAR consists of the IFNAR1 and IFNAR2 heterodimeric subunits, and deletion of either IFNAR1 or IFNAR2 eliminates IFN-I signaling, abolishing its downstream effects. After any IFN-I family member binds, the intracellular subunits of IFNAR1 and IFNAR2 bind to the tyrosine kinases Tyk2 and JAK1, respectively, activating them. These kinases then phosphorylate members of the signal transducer and activator of transcription (STAT) protein family, primarily STAT1 and STAT2. These phosphorylated STATs then form STAT1 or STAT2 homodimers or STAT1/STAT2 heterodimers. These dimers then combine with IRF9 to form the ISGF3 transcription factor, which enters the nucleus and binds to interferon stimulated response elements (ISREs), inducing the transcription and subsequent translation of interferon stimulated genes (ISGs).
In a previous study, we showed that the IFN-I signaling inhibitor USP18, one of the most highly upregulated ISGs, is largely responsible for the pro-bacterial effect of IFN-I [20]. Deletion of this gene in CD11c+ cells reduced bacterial titers in comparison to WT mice despite strong IFN-I signaling, indicating that the pro-bacterial effects of IFN-I are driven by its own negative regulator in CD11c+ cells. In the current study, we looked upstream of USP18 to investigate whether the pro-bacterial effects of IFN-I signaling are driven by one subtype of IFN-I or by IFN-I signaling more generally. Specifically, we aimed to dissect the roles of IFN-α and IFN-β in the progression of L.m., as differences in their biologic effects during L.m. had not yet been elucidated. Additionally, we sought to determine if IFN-I signal mediation by STAT1 or STAT2 and their associated hetero- and homodimers was more important for altered induction of ISGs by IFN-α and IFN-β in the context of L.m. infection.
Materials and Methods
Mice, Bacteria
C57BL6/J, IFN-β–/–, Stat1–/–, Stat2–/–, Usp18fl/fl and Usp18fl/fl x CD11c-Cre male and female mice (8-12 weeks of age) were used. Mice were maintained in pathogen-free conditions and handling conforms to the requirements of the National Institutes of Health and the Scripps Research Institute Animal Research Committee. L. monocytogenes (ATCC strain 43251) bacteria were a kind gift from Klaus Pfeffer (Institute of Medical Microbiology and Hospital Hygiene, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany) and were maintained in brain heart infusion agar. For all experiments, L.m was injected intravenously at the indicated doses per mouse.
ELISA
IFN-α and IFN-β were measured in the serum according to manufacturer’s protocol (PBL assay science).
Cluster Assay
For quantification of bacteria, organs were homogenized and titrated on brain heart agar plates, Colonies were counted 1-day post-plating.
Antibody treatment
Mice were treated intraperitoneally with 1 mg of anti-IFNAR-1 clone MAR1-5A3 (Leinco; MO, USA), 1 mg of anti-IFN-α clone TIF-3C5 (Leinco), 500µg of anti-IFN-β clone HDß-4A7 (Leinco), or isotype IgG1 (Leinco).
Generation of bone marrow–derived-dendritic cells and transfer experiments
Bone marrow was harvested from murine femurs and tibias. Erythrocytes were lysed with ACK lysis buffer, and dendritic cells were generated by culturing bone marrow cells in VLE-DMEM (Sigma, MO, USA) including 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher; MA, USA), 0.1% (v/v) β-mercaptoethanol (β-ME) (Invitrogen, CA, USA), and 10 ng/ml granulocytes macrophage colony-stimulating factor (GM-CSF) (made in-house).
Total RNA extraction, c-DNA synthesis, and quantitative real-time PCR
RNA was isolated from splenocytes or sorted cells with TRIzol (Thermo Fisher) as described in the manufacture’s protocol. The RNA was reverse-transcribed into cDNA with the Quantitect Reverse Transcription Kit (Qiagen, Germany). Gene expression analysis was performed with assays from Qiagen: glyceraldehyde 3-phosphate dehydrogenase (GAPDH; QT01658692), Ifn-α4 (QT01774353), Ifn-β1 (QT00249662), Stat1 (QT00162183), Stat2 (QT00160216), Irf7 (QT00245266), Prkr (QT00162715), Usp18 (QT00167671), Ifnar2 (QT00102340), Oas1 (QT01056048), and Isg15 (QT02274335). Relative quantities (RQs) were determined with the equation RQ = 2–ddCt.
Statistics
Data are expressed as mean ± S.E.M. Unpaired two-tailed student’s t-tests were calculated using GraphPad Prism software to detect statistically significant differences between groups. Significant differences between several groups were detected by two-way analysis of variance (ANOVA). The level of statistical significance was set at: n.s. not significant, P* < 0.05, P** < 0.01, P*** < 0.001 or P**** < 0,0001.
Results
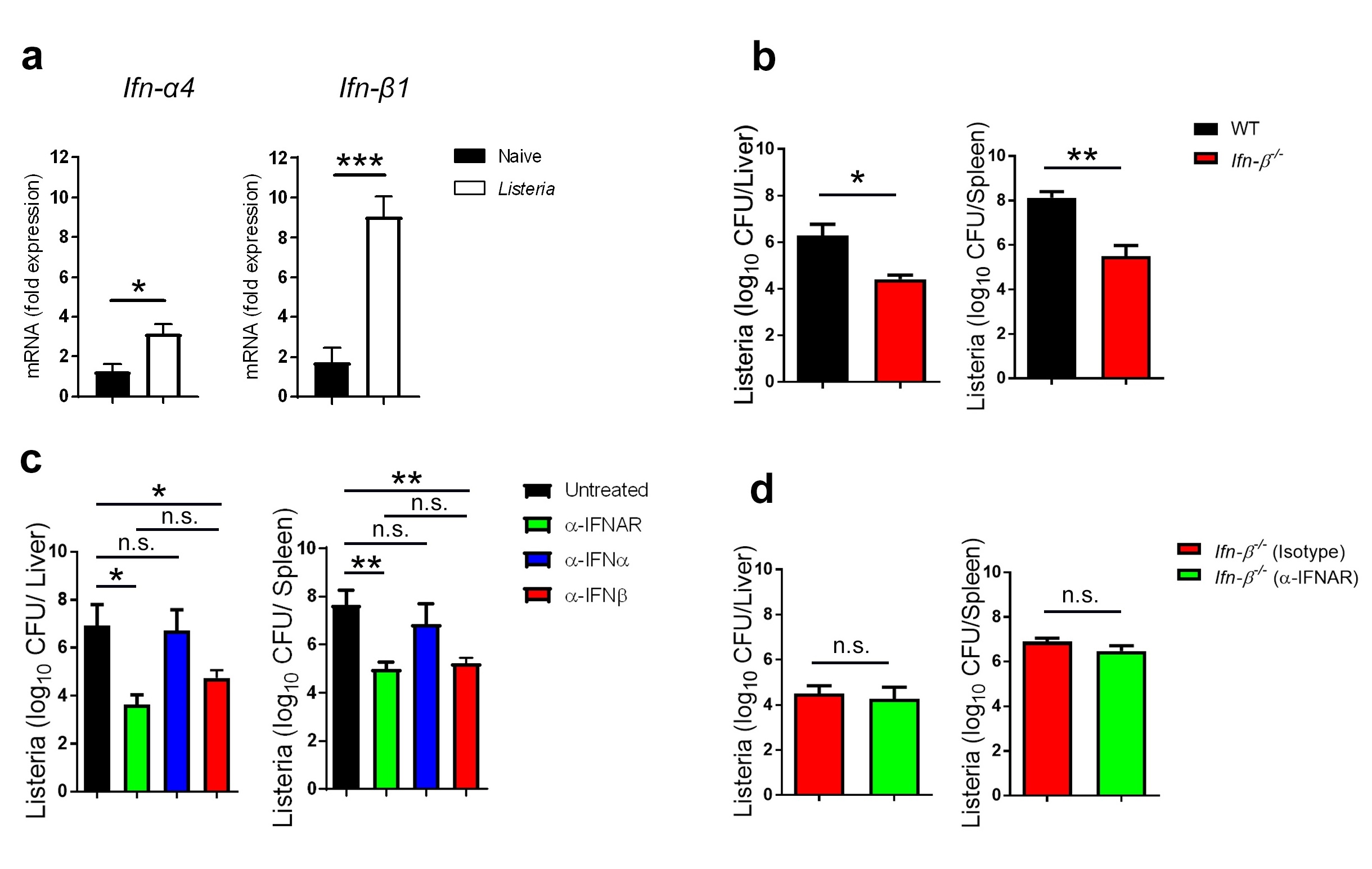
Neutralization of IFN-β signaling enhances bacterial control
First, we measured IFN-I subtype production after infection with Listeria monocytogenes (L.m.). Specifically, we focused on expression of IFN-β, as it is the only IFN-β type, and IFN-α4, which has been shown to be induced in an initial wave of transcription which relies on
IRF3 [21, 22]. We analyzed IFN-I gene expression in the spleen and found that the expression of both Ifn-α4 and Ifn-β1 were elevated 24h after L.m. infection (Fig. 1a). Interestingly, we observed a larger upregulation of Ifn-β1 than Ifn-α4 transcripts (Fig. 1a). Because IFN-β was the most upregulated IFN-I, we asked whether mice lacking IFN-β (IFN-β-/- mice) better control L.m. We found that IFN-β–/– mice exhibit significantly lower titers of L.m. than control mice in both liver and spleen tissue (Fig. 1b). In order to better compare the differential effects of IFN-α and IFN-β during L.m. infection, we treated mice either with IFN-α or IFN-β neutralizing antibodies. As a comparison, we treated another group with an IFNAR blocking antibody to ablate all IFN-I signaling. Interestingly, mice which received an IFN-α-neutralizing antibody which neutralizes six IFN-α species (IFN-Α1, -α4, -α5, -α11, and -α13) [23] had similar bacterial titers to control mice (Fig. 1c), whereas groups treated with anti-IFNAR or anti-IFN-β antibodies exhibited lower bacterial titers than control mice, with both groups controlling the infection equally well (Fig. 1c). Together, these data suggest that IFN-β is the principal driver of the pro-bacterial effect of IFN-I signaling. To further confirm this finding, we treated IFN-β–/– mice with anti-IFNAR antibody or isotype control antibody prior to infection with L.m. We found that both groups showed similar bacterial titers, suggesting that the anti-bacterial effects associated with IFNAR blockade are due to disruption of IFN-β, with minimal requirements for IFN-α signaling (Fig. 1d).
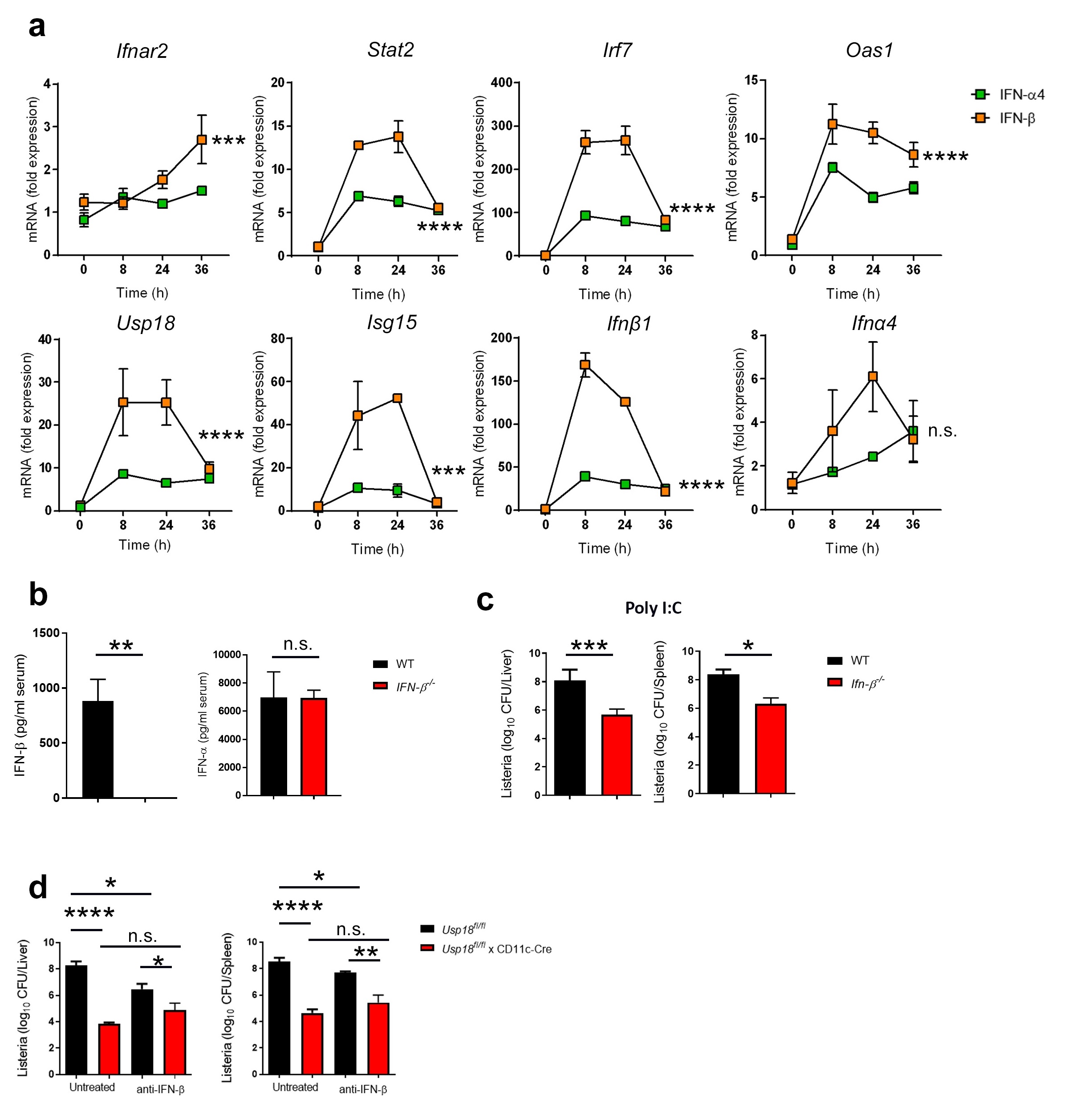
IFN-β induces higher ISG expression than IFN-α in dendritic cells
Next, we wanted to investigate mechanistically why IFN-β, but not IFN-α, induces pro-bacterial effects. Because IFN-β has a greater affinity for IFNAR [19] we speculated that IFN-β induces stronger intracellular IFN signaling than IFN-α. To test this, we generated dendritic cells (DCs) from bone marrow precursors, as DCs must be infected by L.m. for successful dissemination [24] and because deletion of IFNAR specifically from DCs abolishes the pro-bacterial effects of IFN-I [20]. We treated DCs with equal amounts of either IFN-β or IFN-α4 and, at different time points, we measured interferon stimulated gene (ISG) induction by qRT-PCR. We found that ISG expression was significantly higher in IFN-β-treated DCs than IFN-α4-treated DCs (Fig. 2a). Specifically, we found that IFN-β treatment induced an over 100-fold increase in expression of the IFN-Β gene while generating only a modest induction of IFN-α4. Conversely, IFN-α4 treatment induced minimal changes to both IFN-β and IFN-α4 gene expression in comparison to IFN-β treatment (Fig. 2a). This indicates that IFN-β but not IFN-α can feed the IFN-β positive induction loop. To further interrogate this, we treated WT and IFN-β‑/‑ mice with the TLR3 agonist Poly I:C. Such treatment in WT mice leads to the elevated production of both IFN-α and IFN-β [25]. Interestingly, the absence of IFN-β has no impact on the production of IFN-α (Fig. 2b). Moreover, deletion of IFN-β resulted in a significant reduction of bacterial loads following Poly I:C treatment despite similar levels of IFN-α (Fig. 2 c), indicating that IFN-β is primarily responsible for elevated bacterial replication in scenarios with maximal IFN-levels. Together, our data strongly suggests that IFN-β, but not IFN-α, feeds an IFN-Β positive feedback loop that promotes L.m infection.
In our previous study, we showed that, among all ISGs, USP18 is the dominant gene responsible for the pro-bacterial effect of IFN-I [20]. Specifically, we found that USP18 expressed in CD11c+ cells drive this effect. We noticed an early and significant increase in Usp18 expression in dendritic cells following IFN-β treatment compared to IFN-α4 treatment (Fig. 2a). We next asked if CD11c+ cell-intrinsic deletion of USP18 would abolish the pro-bacterial effects of IFN-β. To do this, we infected Usp18fl/fl and Usp18fl/fl x CD11c-Cre mice with L.m. in the presence or absence of IFN-Β neutralizing antibody. Interestingly, mice lacking USP18 in CD11c+ cells showed similar bacterial titers to anti-IFN-β treated littermate controls (Fig. 2d). These results indicate that IFN-β is the dominant upstream inducer of USP18 in CD11c+ cells. Thus, IFN-β increases susceptibility to L.m. due to its ability to induce elevated levels of USP18 expression.
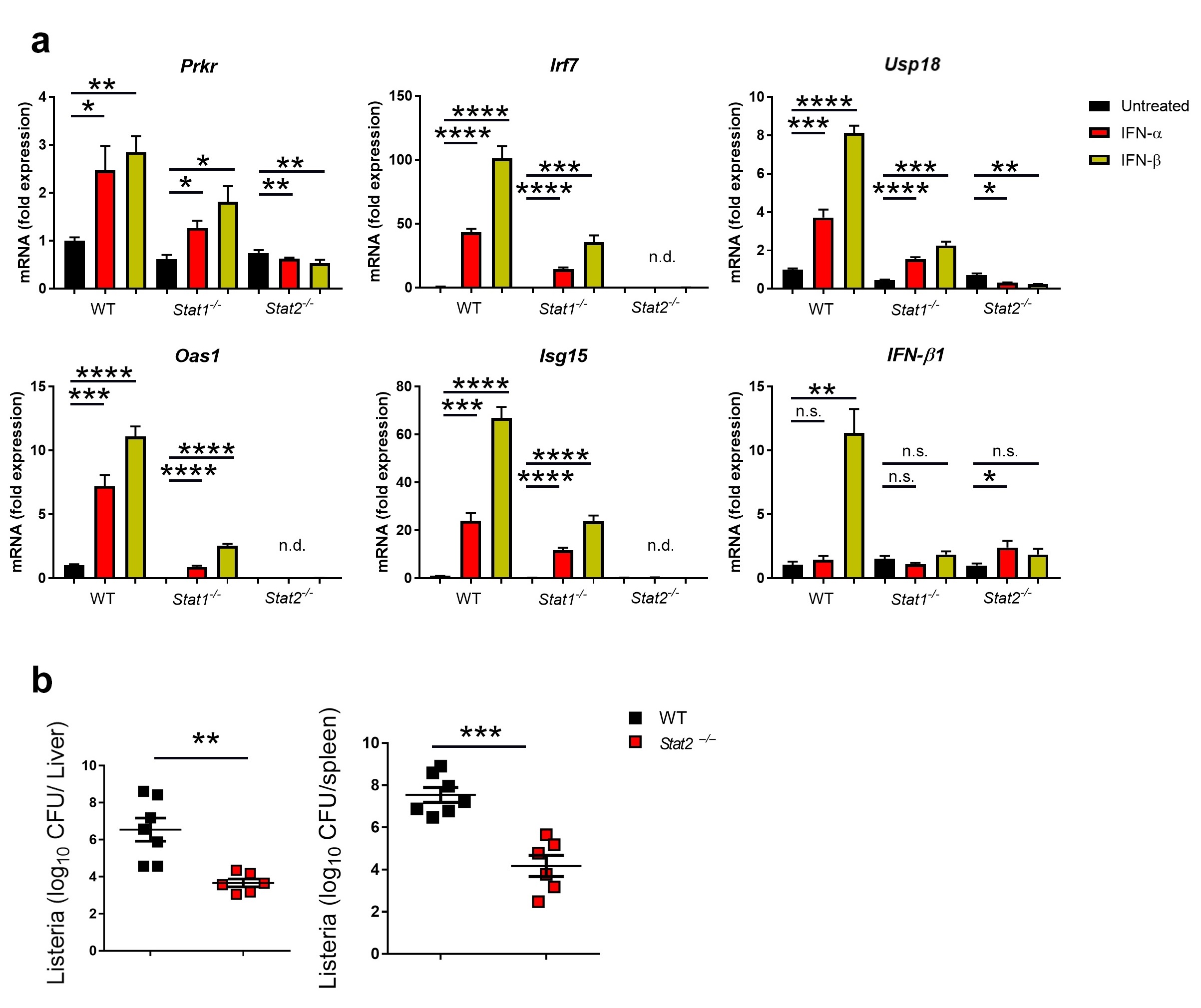
STAT2 is the dominant transducer of pro-Listeria IFN-β signals
Because IFN-β induces USP18 more strongly than IFN-α, we hypothesized that IFN-β signals may be transduced differently than IFN-α. To test this, we asked whether IFN-β signaling post-binding to IFNAR is predominately mediated through STAT1/STAT1 homodimers, STAT2/STAT2 homodimers, or STAT1/STAT2 heterodimers. To investigate this, we generated DCs from the bone marrow of WT, Stat1–/– and Stat2–/– mice and treated them with either IFN-α or IFN-β or left them untreated. We found that the absence of either Stat1 or Stat2 blunted the expression of ISGs relative to WT DCs treated with IFN-I, with STAT2-deficiency blunting ISG expression more than STAT1-deficiency (Fig. 3a). This suggests that both subtypes of IFN-I require both STAT1 and STAT2 for maximal signaling (Fig. 3a), which is consistent with the known signaling pathway of IFN-I. Interesting, this data also suggests that, in DCs, STAT2 is the dominant signal transducer for both IFN-I subtypes. To investigate whether lack of STAT2 alone is sufficient to inhibit the pro-bacterial effect of IFN-β, we infected WT mice and Stat2–/– mice with L.m. and measured bacterial titers in different organs. Indeed, Stat2–/– mice were more resistant to L.m. infection in comparison to WT mice (Fig. 3b), suggesting that, at a minimum, dimers involving STAT2, be them STAT1/STAT2 heterodimers or STAT2/STAT2 homodimers, are required for mediating the pro-bacterial effects of IFN-β signaling.
Discussion
Although IFN-I signaling has been known to play a pro-bacterial role, the IFN-I subtype most responsible for this effect was unknown. In this work, we show for the first time that, among members of IFN-I family, IFN-β is the main IFN-I subtype responsible for the pro-bacterial effect of IFN-I. We show that IFN-β induces higher expression of ISGs than IFN-αs, including USP18. As part of this work, we confirm in DCs that IFN-β signaling is dominantly mediated by STAT2-containing dimers. Because the canonical IFN-I signaling pathway involves STAT1/STAT2 heterodimers or STAT2/STAT2 homodimers combining with IRF9 to form ISGF3 or ISFG3-like complexes [26] this suggests IFN-β signals are transduced through this canonical pathway, and that this canonical signaling results in the pro-bacterial effects of IFN-I. Furthermore, our results suggest that potential non-canonical signaling through STAT1/STAT1 homodimers, which can bind to gamma activated sequences (GASs), cannot compensate for lack of STAT2-mediated signaling in the context of L.m. infection. Our results are further supported by a study demonstrating that STAT1-/- mice display elevated bacterial loads and increased mortality following L.m infection [27], demonstrating STAT1 signaling is necessary for control of L.m.
Furthermore in this report, we show that IFN-β is able to induce further IFN-β expression in a positive feedback loop. This positive induction of IFN-β may explain why IFN-β has an overall stronger effect than other IFN-I subtypes. Despite the fact that all members of the IFN-I family share the same receptor, as IFN-β signaling induces even more IFN-β, resulting in an additive signaling effect. In contrast, IFN-α4 treatment did not induce IFN-α nor IFN-β in DCs. Additionally, we found that this IFN-β self-induction is STAT2-dependent. Previous studies showed that STAT2 can form complexes together with IRF9 and bind ISRE sequences [28-30], and our IFN-β upregulation data suggests that a unique subset of ISGs can be induced depending on the specific STAT complexes induced by certain IFN-I subtypes.
Previously, we showed that USP18 plays a deleterious role during L.m. infection. We reported that one explanation for this phenotype was the ability of USP18 to inhibit antibacterial effects of TNF-α. Nevertheless, other mechanisms may be involved. Here, we show that STAT2-deficient cells exhibit lower USP18 expression, correlating with lower bacterial titers in Stat2–/– mice, suggesting that altered STAT signaling induced by IFN-β is the primary signaling mediator driving the pro-bacterial effect of USP18. Further studies are needed to identify IFN-β blockade as a potential clinical treatment for bacterial infections in humans. In conclusion, we define IFN-β as the principal IFN-I member that enhances bacterial replication during L. monocytogenes infection.
We want to thank our technician Melissa Wilson, Jessica Van Leeuwen from the Scripps Department of Animal Resources, and the FACS Core facility staff at Scripps Research Institute for technical support.
Funding Sources
N.S. was supported by a Deutsche Forschungsgemeinschaft (DFG) fellowship (SH 1140/2-1).
The authors declare that they have no financial interests that relate to the research described in this paper.
| 1 Davidson S, Crotta S, McCabe TM, Wack A: Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun 2014;5:3864. https://doi.org/10.1038/ncomms4864 |
||||
| 2 Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H: Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011;146:980-991. https://doi.org/10.1016/j.cell.2011.08.015 |
||||
| 3 Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB: Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013;340:207-211. https://doi.org/10.1126/science.1235214 |
||||
| 4 Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG: Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013;340:202-207. https://doi.org/10.1126/science.1235208 |
||||
| 5 Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, Sheehan KC, Schreiber RD, Oldstone MB: Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 2015;17:653-661. https://doi.org/10.1016/j.chom.2015.04.005 |
||||
| 6 Davidson S, Maini MK, Wack A: Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J Interferon Cytokine Res 2015;35:252-264. https://doi.org/10.1089/jir.2014.0227 |
||||
| 7 Decker T, Muller M, Stockinger S: The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol 2005;5:675-687. https://doi.org/10.1038/nri1684 |
||||
| 8 Boxx GM, Cheng G: The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe 2016;19:760-769. https://doi.org/10.1016/j.chom.2016.05.016 |
||||
| 9 O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G: Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 2004;200:437-445. https://doi.org/10.1084/jem.20040712 |
||||
| 10 Navarini AA, Recher M, Lang KS, Georgiev P, Meury S, Bergthaler A, Flatz L, Bille J, Landmann R, Odermatt B, Hengartner H, Zinkernagel RM: Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc Natl Acad Sci U S A 2006;103:15535-15539. https://doi.org/10.1073/pnas.0607325103 |
||||
| 11 Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA: Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 2004;200:527-533. https://doi.org/10.1084/jem.20040976 |
||||
| 12 Carrero JA, Calderon B, Unanue ER: Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol 2004;172:4866-4874. https://doi.org/10.4049/jimmunol.172.8.4866 |
||||
| 13 Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, Ho L, Sauer JD, Iwakura Y, Metzger DW, Monack DM: Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol 2010;184:3755-3767. https://doi.org/10.4049/jimmunol.0902065 |
||||
| 14 Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL: Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med 2010;207:327-337. https://doi.org/10.1084/jem.20091746 |
||||
| 15 Pestka S, Krause CD, Walter MR: Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004;202:8-32. https://doi.org/10.1111/j.0105-2896.2004.00204.x |
||||
| 16 Diaz MO, Pomykala HM, Bohlander SK, Maltepe E, Malik K, Brownstein B, Olopade OI: Structure of the human type-I interferon gene cluster determined from a YAC clone contig. Genomics 1994;22:540-552. https://doi.org/10.1006/geno.1994.1427 |
||||
| 17 Hoofnagle JH, Mullen KD, Jones DB, Rustgi V, Di Bisceglie A, Peters M, Waggoner JG, Park Y, Jones EA: Treatment of chronic non-A,non-B hepatitis with recombinant human alpha interferon. A preliminary report. N Engl J Med 1986;315:1575-1578. https://doi.org/10.1056/NEJM198612183152503 |
||||
| 18 Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, Fischer JS, Goodkin DE, Granger CV, Simon JH, Alam JJ, Bartoszak DM, Bourdette DN, Braiman J, Brownscheidle CM, Coats ME, Cohan SL, Dougherty DS, Kinkel RP, Mass MK, et al.: Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol 1996;39:285-294. https://doi.org/10.1002/ana.410390304 |
||||
| 19 Piehler J, Thomas C, Garcia KC, Schreiber G: Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev 2012;250:317-334. https://doi.org/10.1111/imr.12001 |
||||
| 20 Shaabani N, Honke N, Nguyen N, Huang Z, Arimoto KI, Lazar D, Loe TK, Lang KS, Prinz M, Knobeloch KP, Zhang DE, Teijaro JR: The probacterial effect of type I interferon signaling requires its own negative regulator USP18. Sci Immunol 2018;3:eaau2125. https://doi.org/10.1126/sciimmunol.aau2125 |
||||
| 21 McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A: Type I interferons in infectious disease. Nat Rev Immunol 2015;15:87-103. https://doi.org/10.1038/nri3787 |
||||
| 22 Lin R, Mamane Y, Hiscott J: Multiple regulatory domains control IRF-7 activity in response to virus infection. J Biol Chem 2000;275:34320-34327. https://doi.org/10.1074/jbc.M002814200 |
||||
| 23 Sheehan KC, Lazear HM, Diamond MS, Schreiber RD: Selective Blockade of Interferon-alpha and -beta Reveals Their Non-Redundant Functions in a Mouse Model of West Nile Virus Infection. PLoS One 2015;10:e0128636. https://doi.org/10.1371/journal.pone.0128636 |
||||
| 24 Neuenhahn M, Kerksiek KM, Nauerth M, Suhre MH, Schiemann M, Gebhardt FE, Stemberger C, Panthel K, Schroder S, Chakraborty T, Jung S, Hochrein H, Russmann H, Brocker T, Busch DH: CD8alpha+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity 2006;25:619-630. https://doi.org/10.1016/j.immuni.2006.07.017 |
||||
| 25 Alexopoulou L, Holt AC, Medzhitov R, Flavell RA: Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001;413:732-738. https://doi.org/10.1038/35099560 |
||||
| 26 Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR: A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front Immunol 2018;9:1135. https://doi.org/10.3389/fimmu.2018.01135 |
||||
| 27 Kernbauer E, Maier V, Stoiber D, Strobl B, Schneckenleithner C, Sexl V, Reichart U, Reizis B, Kalinke U, Jamieson A, Muller M, Decker T: Conditional Stat1 ablation reveals the importance of interferon signaling for immunity to Listeria monocytogenes infection. PLoS Pathog 2012;8:e1002763. https://doi.org/10.1371/journal.ppat.1002763 |
||||
| 28 Abdul-Sater AA, Majoros A, Plumlee CR, Perry S, Gu AD, Lee C, Shresta S, Decker T, Schindler C: Different STAT Transcription Complexes Drive Early and Delayed Responses to Type I IFNs. J Immunol 2015;195:210-216. https://doi.org/10.4049/jimmunol.1401139 |
||||
| 29 Majoros A, Platanitis E, Szappanos D, Cheon H, Vogl C, Shukla P, Stark GR, Sexl V, Schreiber R, Schindler C, Muller M, Decker T: Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1 EMBO Rep 2016;17:367-382. https://doi.org/10.15252/embr.201540726 |
||||
| 30 Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, Decker T: Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front Immunol 2017;8:29. https://doi.org/10.3389/fimmu.2017.00029 |
||||