×
![]()
Corresponding Author: Dmitriy Minond
Rumbaugh-Goodwin Institute for Cancer Research, Nova Southeastern University, 3321 College Avenue, CCR 6th Floor, Fort Lauderdale, FL 33314 (USA)
E-Mail dminond@nova.edu
Development and Pilot Screen of Novel High Content Assay for Down Regulators of Expression of Heterogenous Nuclear Ribonuclear Protein H2
Juan Dieza Sumitha Rajendraraob Shadi A. Baajourc Praathibha Sripadhana Timothy P. Spicerb Louis D. Scampaviab Dmitriy Minonda,c
aRumbaugh-Goodwin Institute for Cancer Research, Nova Southeastern University, Fort Lauderdale, FL, USA, bThe Scripps Research Molecular Screening Center, Department of Molecular Medicine, Scripps Research, Jupiter, FL, USA, cDr. Kiran C. Patel College of Allopathic Medicine, Nova Southeastern University, Fort Lauderdale, FL, USA
Introduction
There is an unmet need for the discovery of new molecules that can be targeted to induce melanoma cell-specific death. Several decades of research into kinase inhibitors and immunotherapy have led to the development of a number of new drugs along with the resulting extension of patient lives, but resistance often occurs and it appears that multiple mechanisms need to be targeted in order to provide sustained disease control [1, 2].
Our recent report suggests that spliceosomal protein heterogeneous nuclear ribonucleoprotein H2 (hnRNPH2) can be targeted to induce melanoma-specific cell death that can address an unmet need for such novel therapeutic approaches focused on novel mechanisms [3].
Targeting multiple pathways may help prevent or overcome drug resistance [4-6]. Modulation of hnRNPH2 induces an autophagy which was shown to be an effective strategy in overcoming multi-drug resistance (MDR) in cancers [7]. Autophagy plays a role in the development as well as overcoming drug resistance in melanoma [8], therefore, an ability to shift balance from pro- to anti-resistance can also be beneficial. Because of this ability, hnRNPH2 modulation may also address the lack of therapies for NRAS mutants and other subtypes of melanoma. Autophagy also regulates cytokine secretion and antigen presentation and, therefore, contributes to anti-tumor immune responses [9-11].
Based on these considerations, modulation of hnRNPH2 may overcome drug resistant melanoma and help prevent occurrence of resistance, which would be a significant improvement over existing drugs such as vemurafenib.
With this in mind, we decided to explore existing chemical diversity in order to identify novel down regulators of hnRNPH2. Typically, this requires an assay suitable to compound screening in a high throughput format (HTS) [12]. However, we were not able to identify an assay platform that would allow us to do so, therefore, we decided to develop such a platform. Herein we present the results of HTS-amenable assay development and its validation.
Materials and Methods
WM266-4-H2GFP (Green Fluorescent Protein) cell line establishment
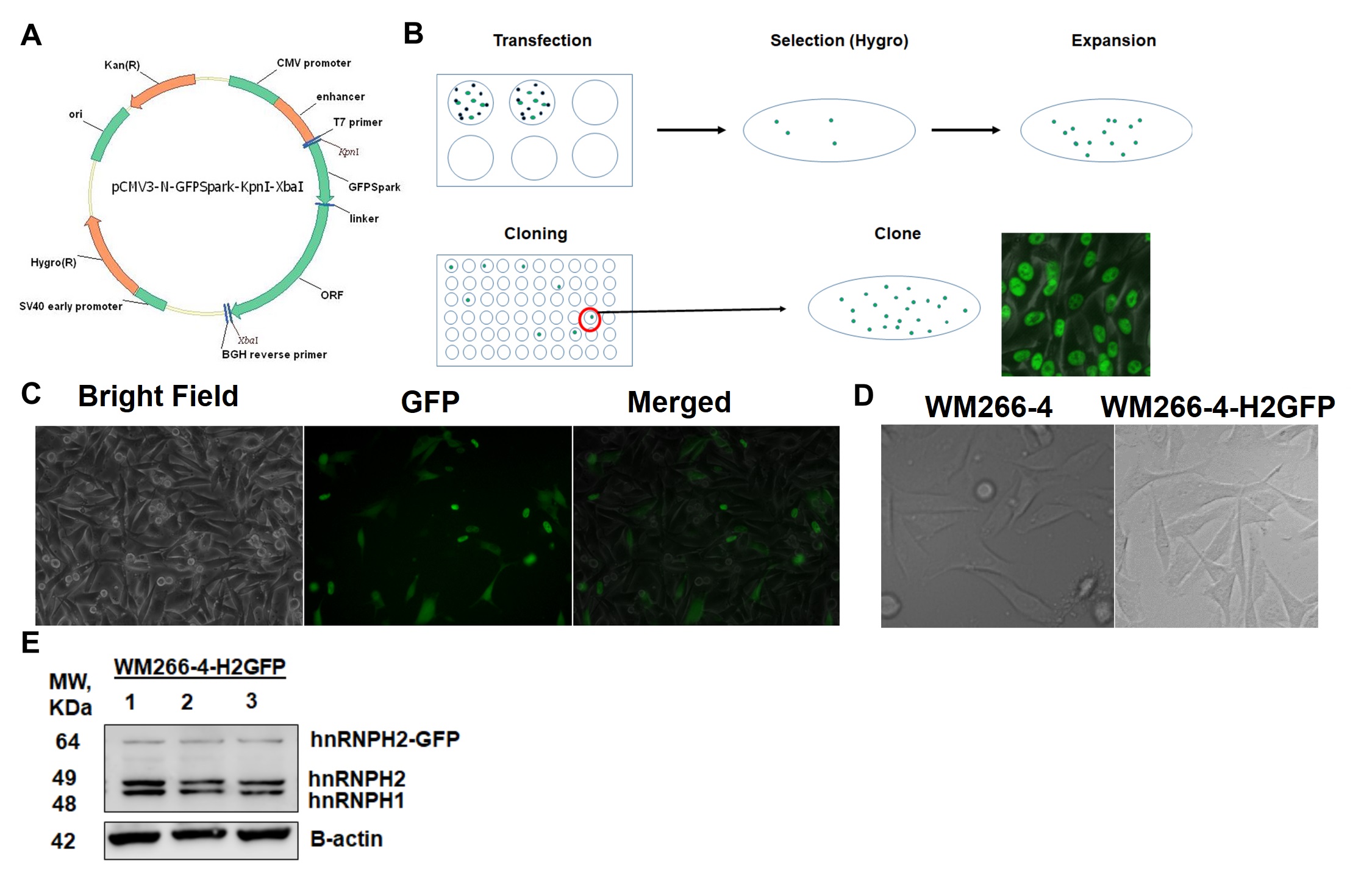
WM266-4 cells [13] were purchased from Rockland Immunochemicals, Inc. (Limerick, PA) and authenticated by the vendor. Subsequent authentications and mycoplasma certification was done at Johns Hopkins University Genetic Resources Core Facility. WM266-4 cells were plated in 6-well plate at 5 x 105 cells/well. After cells were attached (next day) they were transfected with GFP-H2 plasmid (Sino Biologics) using Lipofectamine Plus according to the manufacturer directions. Briefly, 2 μL of plasmid, 5 μL lipofectamine and 2.5 μL Plus reagent were mixed in 500 μL serum-free EMEM and after 5 min incubation at RT 250 μL were added to each well. Transfection efficiency was assessed by monitoring green fluorescence. When cells were confluent, they were transferred to a T75 flask and after they adhered selection began by adding 200 μg/mL Hygromycin to the medium. As cells began dying, the medium was frequently changed, maintaining the hygromycin concentration. Two weeks later, when colonies were observed, they were transferred to 6-well plate wells, allowed to grow and then diluted and plated at 1 cell/well in 48-well plates. Hygromycin was always maintained to keep the selection pressure. The colony with the strongest and most uniform expression was selected and H2-GFP expression was assessed by fluorescent microscopy and Western blot. Cells from this colony were sorted, using Fluorescence-Activated Cell Sorting (FACS) to further purify the colony.
Primary WM266-4-H2GFP High Content Screening (HCS) assay in 384 well plate format
1,250 WM266-4-H2GFP cells were plated in low volume black sterile tissue culture treated 384-well plates (Greiner cat# 781099) in 10 µL of media. Next day after plating, compounds from Food and Drug Administration (FDA) approved drug library and pharmacological control (dronedarone) were added to the cells (100 nL per well, 10µM final assay concentration) using the 384 channel pin tool (V&P Scientific, San Diego) mounted on ViaFlo 384 (Integra). After overnight incubation at 37°C, 5% CO2 and 95% relative humidity, the plates were imaged using algorithm on CellInsight CX7 (Thermo Scientific). For each well, the Average Total Object Intensity (ATOI) was measured and used as a basis for calculation of statistic parameters (whole plate average ATOI, %CV, Z’) and EC50 values.
Primary WM266-4-H2GFP HCS assay in 1,536 well plate format
750 WM266-4-H2GFP cells were plated in low volume black, clear bottom sterile tissue low base culture treated 1,536-well plates (Aurora cat# 00019326) in 5 µL of media using a Flying Reagent Dispenser (FRD, Aurora Biosciences).
The next day, cells were pinned with 50nL of the pharmacological control compounds mitomycin and dronedarone using a pin tool transfer unit (Kalypsys/GNF). After overnight incubation at 37°C, 5% CO2 and 95% relative humidity, fluorescent images were acquired on CellInsight image reader using 10x objective collecting 4 fields of view per well (485 nm at a fixed exposure time 0.063 s). We implemented the CellInsight’s target activation assay algorithm which was configured for the screen. Background from the raw images was removed by using low pass filter and smoothing parameters. Primary objects were identified using the fixed threshold method set at 65 and the objects touching the image border were excluded. Further, primary objects were validated by defining the object area and intensity of the objects. The output features such as valid object count and the average total intensity were exported for each plate and data analysis was performed using the ratio of valid object count to Average Total Object Intensity.
GFP interference assay
This assay was conducted the same way as a primary HCS assay except for compound incubation time. Briefly, WM266-4-H2GFP cells were plated in 384-well plates (Greiner cat# 781099) in 10 µL of media. Next day after plating, test compounds were added to the cells (100 nL per well) using the 384 channel pin tool (V&P Scientific, San Diego) mounted on ViaFlo 384 (Integra) and fluorescence emission was scanned immediately using microplate reader Synergy H1 (Biotek).
Cell viability assays
Briefly, WM-266-4 and WM266-4-H2GFP cells were plated in 384-well plates in 8 µL of media. Test compounds and dabrafenib (pharmacological assay control) were prepared as 10-point, 1:3 serial dilutions starting at 300 µM, then added to the cells (4 µL per well) using the 384 channel pin tool (V&P Scientific, San Diego) mounted on ViaFlo 384 (Integra). Plates were incubated for 72 h at 37°C, 5% CO2 and 95% relative humidity. After incubation, 4 µL of CellTiter-Glo® (Promega cat#: G7570) were added to each well and incubated for 15 min at room temperature. Luminescence was recorded using a Biotek Synergy H1 multimode microplate reader. Viability was expressed as a percentage relative to wells containing media only (0%) and wells containing cells treated with DMSO only (100%). Three parameters were calculated on a per-plate basis: (a) the signal-to-background ratio (S/B); (b) the coefficient for variation [CV; CV = (standard deviation/mean) x 100)] for all compound test wells; and (c) the Z’-factor (18).
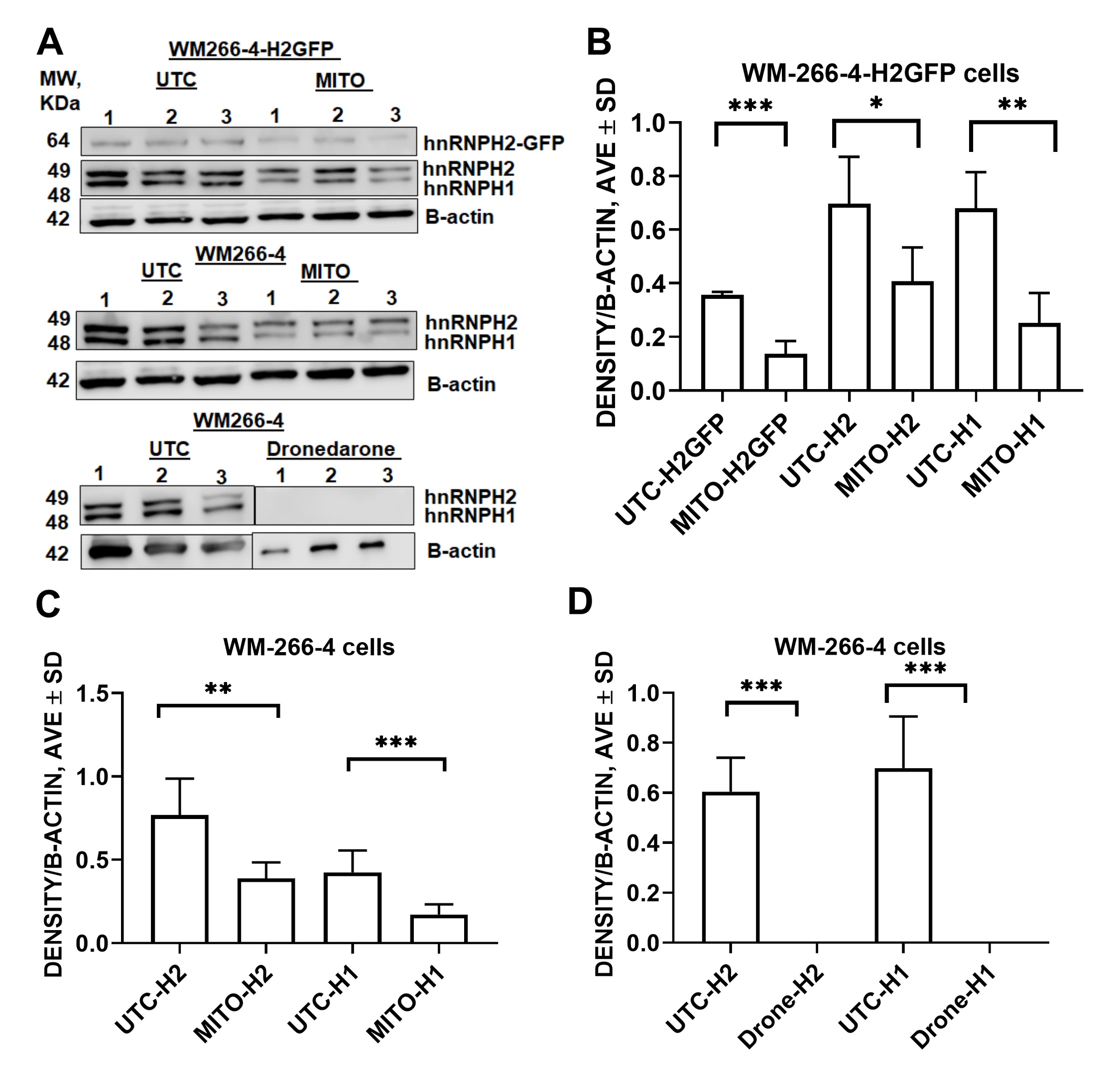
Western blotting for hnRNP H2
WM-266-4 and WM266-4-H2GFP cells were sonicated in RIPA lysis buffer containing protease and phosphatase inhibitors. Protein isolates were subjected to SDS-PAGE followed by transfer to nitrocellulose membrane. hnRNP H2 was detected using rabbit polyclonal antibody (Abgent #: AP13497b; 1:3,000, in 5% milk overnight). After washing with TBST, the membranes were treated with chemiluminescent horseradish peroxidase detection reagent (Thermo Scientific, Cat# 32209) and developed using the Licor imaging system. Licor software was used to quantitate the intensity of protein bands. The protein bands were normalized against loading control (β-actin).
Results
WM266-4-H2GFP cell line establishment
We used a construct encoding for hnRNPH2 tagged with GFPSpark (Sino Biological HG16541-ANG) to stably transfect WM266-4 cells (Fig. 1A). hnRNPH2-GFP was transfected into WM266-4 cells using Lipofectamine LTX (Life Technologies 15338030) according to the manufacturer directions. Transfected WM266-4 cells were selected by culturing them in EMEM supplemented with hygromycin until most of the cells died and some colonies appeared. Those colonies were allowed to grow, collected and cells diluted and plated again in 96-well plates at a dilution of 1 cell per well to obtain clonal populations of the H2-GFP cells (Fig. 1B). After growing these colonies for several weeks, the expression was monitored by fluorescent microscopy and those colonies with the stronger and uniform expression of H2-GFP were selected (Fig. 1C). To further enrich cell population expressing hnRNPH2-GFP, cells were sorted using FACS and WM266-4 cells as non-GFP-expressing control. Resulting WM266-4-hnRNPH2-GFP (H2-GFP) cells showed morphology, viability and doubling rate similar to parental cells (Fig. 1D). Similarly to WM266-4 cells, H2-GFP cells recovered well after LN2 storage. Western blot analysis confirmed expression of H2-GFP (Fig. 1E).
Development of primary HCS assay for hnRNPH2 down regulators
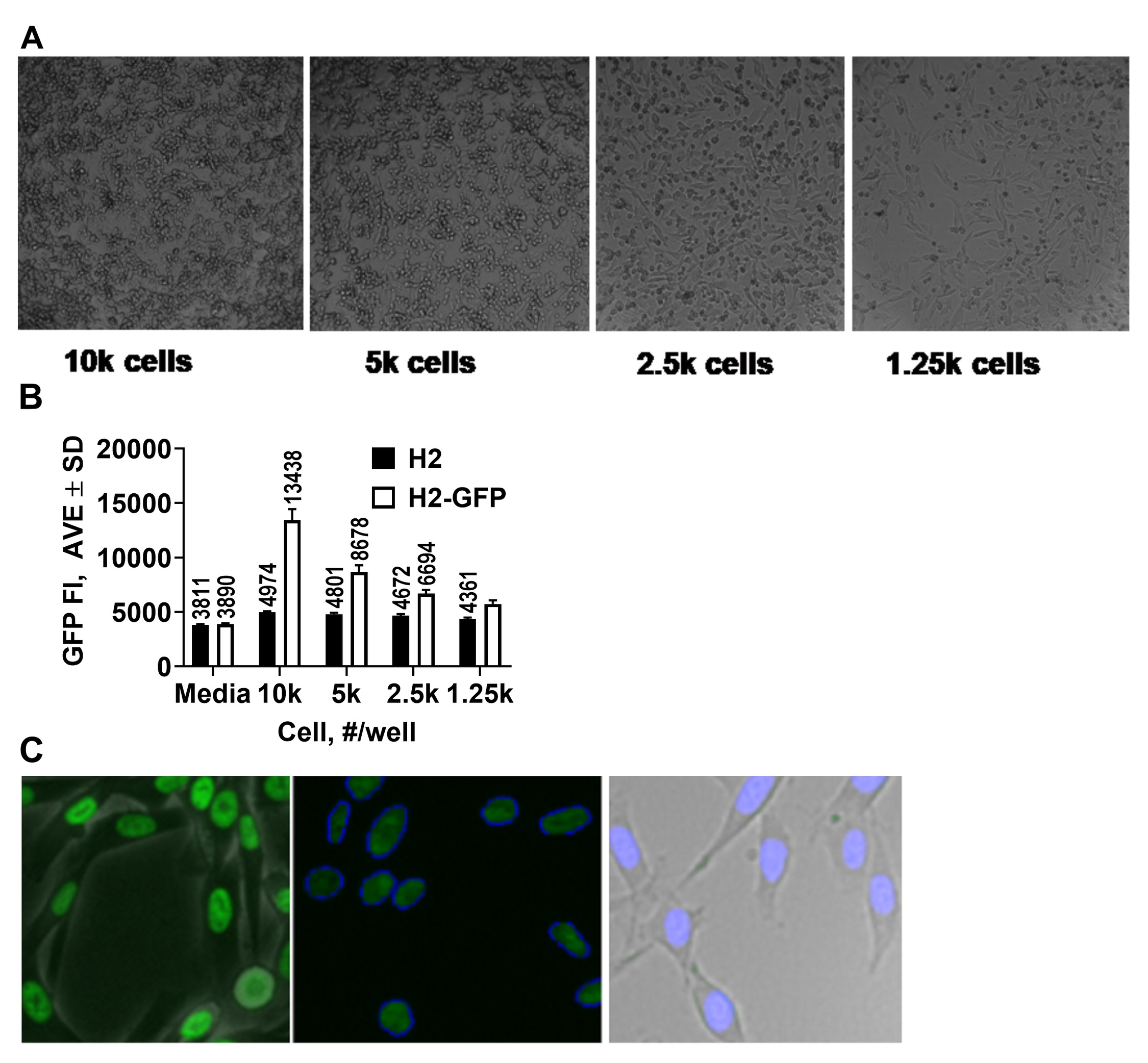
Choice of a readout. GFP fluorescence can be measured in HTS mode by either microplate readers or high content imagers. Microplate readers are preferable for HTS since they are ubiquitous and measurement requires less optimization, whereas high content imagers are more specialized, require fine tuning, and not always available at various HTS facilities. To determine whether an HTS assay can be developed using microplate reader we measured GFP fluorescence of range of WM266-4-H2GFP cells using Nunc® black opaque 384 well plate (Thermo Scientific cat# 264705). As can be seen in Fig. 2A, only 1,250 cells/well resulted in sub-confluent culture, which is a necessary condition for performing biological experiments. Ratio of GFP fluorescence intensity (FI) of GFP and non-GFP WM266-4 cells was 1.3 (Fig. 2B) suggesting that the assay window is very narrow and would not allow for separation of noise and hits. Since reader-based measurement approach did not produce desired signal separation, we proceeded with assay development using high content imager (CellInsight CX7, ThermoFisher Scientific). We utilized black clear-bottom 384 well plates (Greiner cat# 788091). These plates have the same well surface area as Nunc plates that we used for cell number determination (Fig. 2A), therefore, we used 1,250 cells/well of both WM266-4 and WM266-4-H2GFP cells for our experiments. High Content Assays (HCA) rely on algorithms that recognize and draw borders (masks) around objects that need to be quantitated. To enable a GFP fluorescence intensity measurement we developed a method based on a standard algorithm available as a part of CX7 software package. As can be seen in Fig. 2C, our method recognizes nuclei expressing hnRNPH2-GFP very well. This suggested that our algorithm can recognize valid objects and discarding non-specific signals.
Next, we dispensed 1,250 WM266-4-H2GFP cells well in 10μL of EMEM into each well of columns 2-24 of black clear-bottom 384 well plates (Greiner cat# 788091) and 1,250 WM266-4 cells in each well of column 1 and estimated signal-to-basal (S:B) ratio. As expected, wells with WM266-4 cells did not produce a signal while wells with WM266-4-H2GFP cells had Average Total Fluorescence Intensity (ATOI) of 338,408±15382 (n = 368 wells) (Fig. 3A). %CV was 4.5 suggesting great intra-plate signal reproducibility. While we could not calculate a true S:B ratio due to the lack of low ATOI control, the high signal of wells with WM266-4-H2GFP cells (high ATOI control) and a robust %CV suggested that a high content imager can be used to develop an HTS assay.
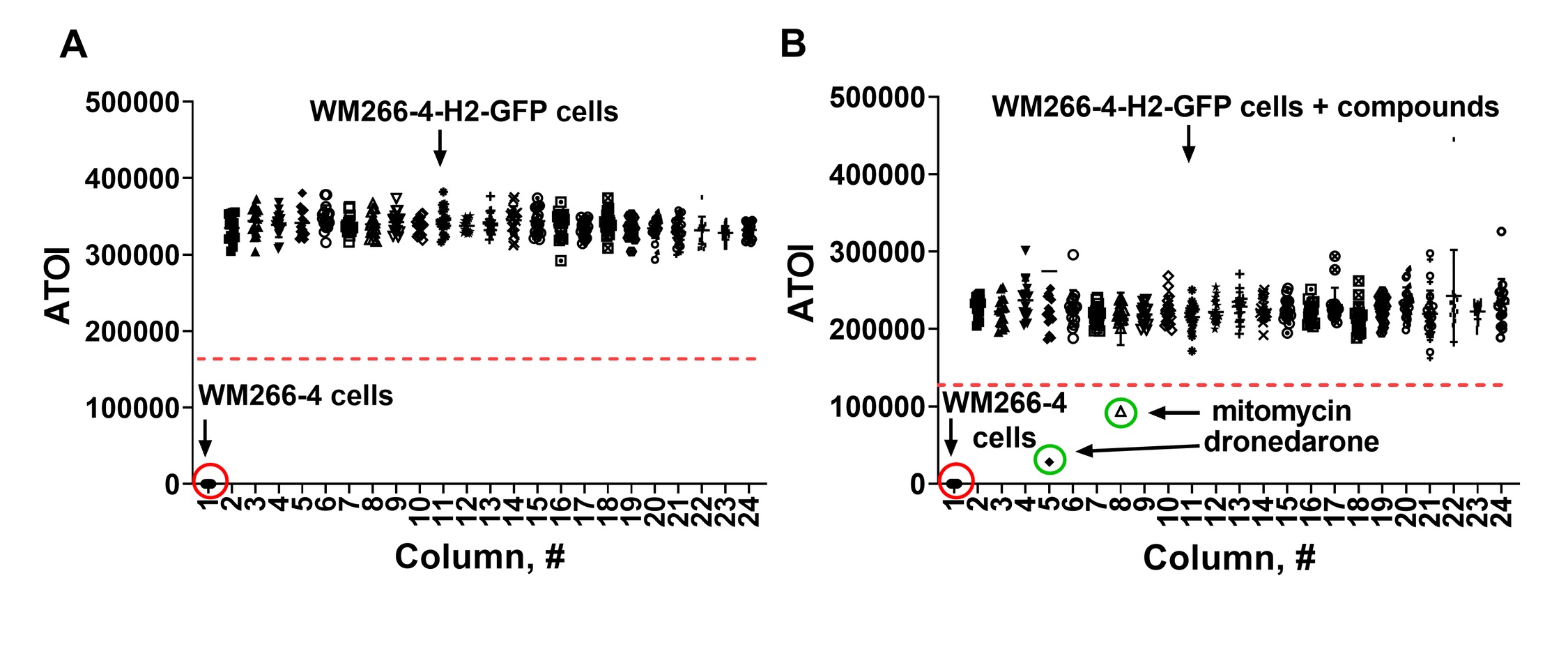
Identification of a pharmacological control for HCS assay. We conducted a screen of FDA-approved drug library (1,400 compounds) to identify a pharmacological control for HCS assay. Assay protocol followed “addition only” format (no extraction, filtration, or washing steps): (1) addition of 1,250 cells in 10 µL of EMEM media with 10% FBS, 1% PenStrep, 50 ug/mL hygromycin, utilizing Integra ViaFlo 384 liquid dispenser, (2) addition of test compounds (10μM final) using pin tool device (VP Scientific), (3) incubation of cells and compounds for 24 hrs at 37°C, 5% CO2 and 95% relative humidity, and (4) measurement of Average Total Fluorescence Intensity using CellInsight CX7 (ThermoFisher) high content imager. We used 50% signal decrease as a hit cutoff (Fig. 3B).
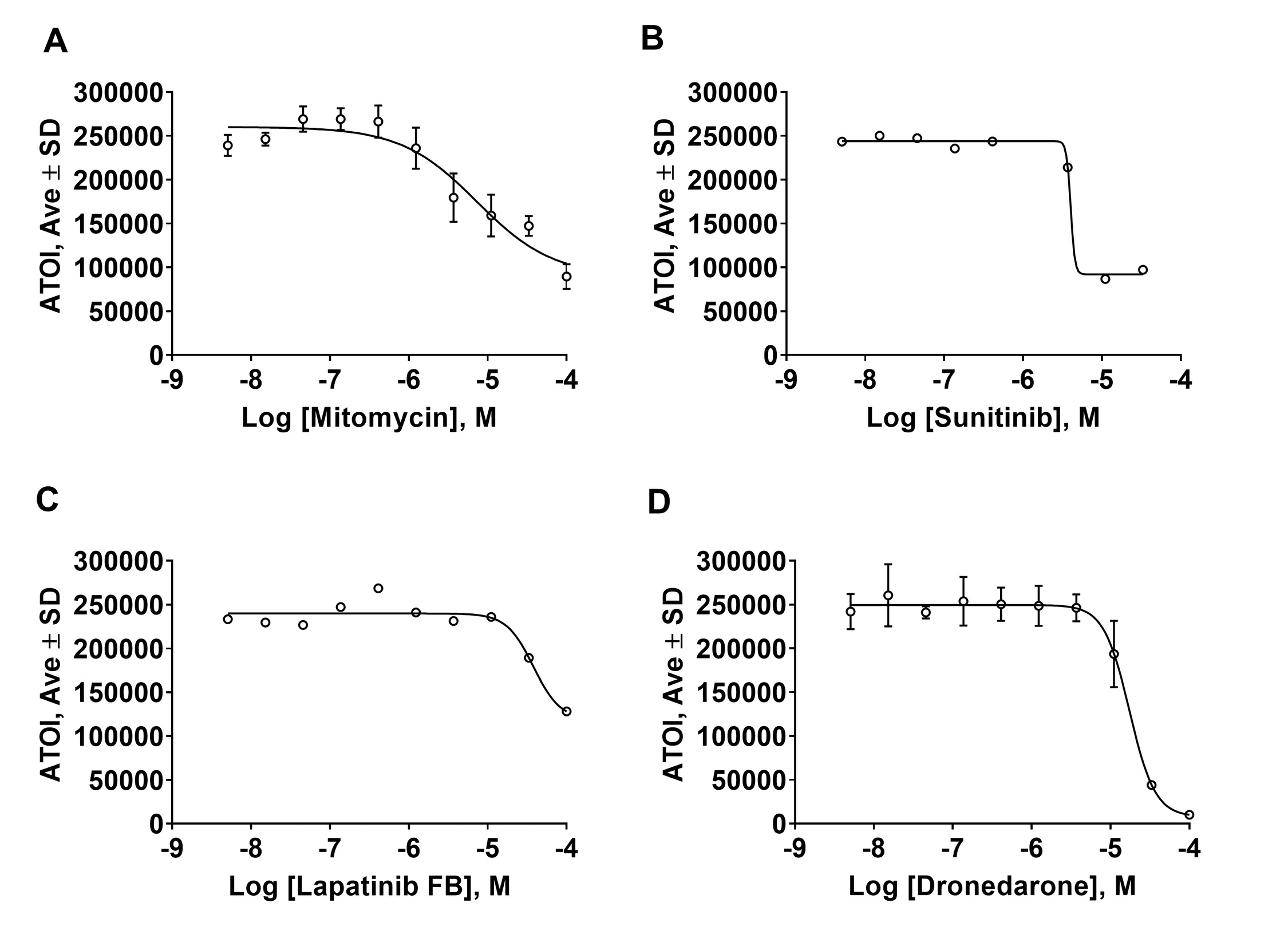
Using this protocol, we found 8 hits (0.5% raw hit rate) of which we were able to purchase six compounds. Out of six commercially available compounds, four confirmed activity in dose response format (0.66% confirmed hit rate), namely dronedarone, mitomycin C, lapatinib free base form, and sunitinib (Fig. 4A-D). Two compounds exhibited EC50 values <20μM (0.07% high activity hit rate) (dronedarone = 17μM and mitomycin = 7.3μM).
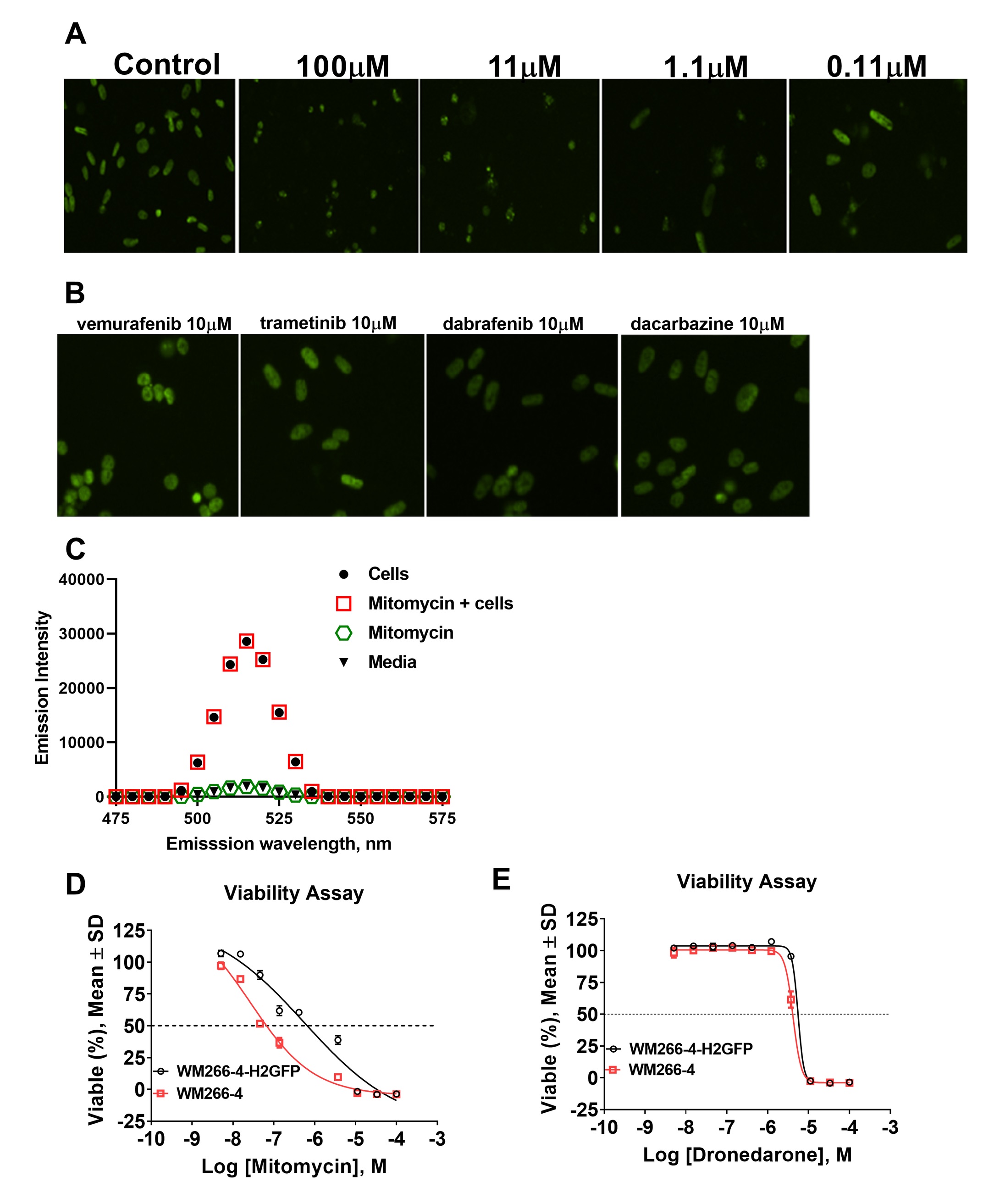
Examination of images of WM266-4-H2-GFP cells in presence of mitomycin showed a dose dependent decrease of fluorescence and nuclei shapes suggesting that HCS assay is capable of correctly recognizing and quantitating the objects in the wells (Fig. 5A). Interestingly, none of FDA approved melanoma drugs (Fig. 5B, trametinib, dabrafenib, vemurafenib, dacarbazine) decreased hnRNPH2 signal further corroborating our hypothesis that hnRNPH2 controls pathways previously unexplored for melanoma therapy [3].
Compounds that absorb light in the GFP emission range (~500nm) could potentially be detected as hits in our assay format, therefore, we examined mitomycin for interference with GFP signal. Fluorescence emission scans of 10µM mitomycin in the presence and absence of WM266-4-H2-GFP cells showed no effect on GFP fluorescence (Fig. 5C) suggesting that mitomycin is a non-artifactual hit.
Next, we evaluated the effect of hits on melanoma cell viability. Interestingly, mitomycin was ~20-fold more potent against WM266-4 cells than WM266-4-H2-GFP cells in viability assay (Fig. 5D, IC50 = 27nM vs 570nM, respectively), whereas dronedarone was equipotent (Fig. 5F, IC50= 5μM). This result suggests different mechanism of action of mitomycin and dronedarone.
Discussion
Our previous studies show that decrease of hnNRPH2 and H1 expression as a result of binding by a small molecule leads to the selective death of melanoma cells [3]. This suggested that hnRNPH2 and associated pathways can be targeted for melanoma therapy; therefore, we decided to expand small molecule compound tools available via HTS effort in order to explore such pathways. We reasoned that compounds that decrease of hnNRPH2 and H1 expression do not necessarily have to bind it. Therefore, the primary biochemical assay that detects just hnNRPH2 binders would be unnecessarily excluding compounds that decrease hnNRPH2 expression via hitherto unknown mechanisms but could be just as valuable. Based on these considerations we developed more physiologically relevant assay that can easily monitor level of hnNRPH2 protein inside the cells in the presence of test compounds.
Most of HTS-amenable detection technologies are based on either luminescence or fluorescence measurements, however, HTS libraries contain ~3% luciferase inhibitors [14, 15] and spectrally active compounds [16]. Since we are interested in compounds that decrease assay signal, luciferase inhibitors would result in ~3% false positives. On other hand, fluorescent compounds could mask some true hits by artificially increasing fluorescence (false negatives) but would not produce false positives. Quenchers of fluorescence are much more rare, therefore, there is less concern about false positives when using fluorescence rather than luciferase reporter. For example, there estimated to be ~0.01-0.2% of compounds with Ex480nm/Em535nm (green fluorescence) [15]. Indeed, none of the pilot screen hits affected GFP fluorescence suggesting that our assay is not prone to fluorescent artifacts possibly due to the high quantum yield of SparkGFP.
The purpose of developing of an assay for hnRNPH2 is to discover novel down regulators of its expression working via different mechanisms. This, in turn, will help researchers to gain an insight into pathways and mechanisms that depend on hnRNPH2 expression. Two confirmed hit compounds discovered as a result of screen of FDA library, dronedarone and mitomycin C, have not been described previously in connection with hnRNPH2 and, therefore, could be valuable chemical tools to study its biology. Mitomycin C is a pro-drug that can be activated intracellularly by several oxido-reductases whose expression is often elevated in cancers [17]. Multiple mechanisms by which mitomycin C kills cancer cells have been proposed, including formation of DNA and RNA adducts [18]. Global expression profiling of U2OSE64b cells treated with mitomycin C showed that it affected mRNA splicing and transport [19], which is consistent with what is known about hnRNPH2 role [20] in cellular processes. Mitomycin is used in clinic for various indications including conjunctival melanoma [21]. Mitomycin was less potent against WM266-4-H2GFP cells which is similar to 2155-14, a known hnRNPH2-binder [3], which exhibits the same trend in inhibiting of WM266-4 and WM266-4-H2GFP cells (Fig. 1E).
Dronedarone is a multi-ion channel blocker used in the clinic as an anti-arrhythmic medicine [22]. It was active against ovarian [23] and breast cancer cells in vitro and in vivo [24], however, its activity against melanoma was not previously demonstrated. However, another calcium channel blocker, amlodipine, inhibited uveal malignant melanoma and cutaneous malignant melanoma cell lines with IC50 values of 13.1 µM and 15.9 µM,
respectively [25], which is similar to dronedarone’s potency (17±1.5μM) against cutaneous malignant melanoma (WM266-4). Based on dronedarone’s equipotent inhibition of
WM266-4 and WM266-4-H2GFP cells (Fig. 5E) it is reasonable to hypothesize that its effect is not modulated by hnRNPH2 binding. It is also possible that hnRNPH2 down regulation is a consequence of ion channel blockade by dronedarone.
As discussed above, both biochemical and cell-based HTS assays have well known limitations based on the method of detection (e.g., fluorescence). Additionally, one specific limitation of cell-based assays is an inability to differentiate between generally cytotoxic compounds (e.g., membrane disruptors) and compounds killing cancer cells via specific targets. While dronedarone and mitomycin appear to decrease GFP signal it is possible that it is done not via direct binding of hnRNPH2, but via a different mechanism(s). Despite this limitation these hit compounds provide suitable assay controls that will allow researchers to benchmark assay performance during large scale ultra-high throughput screening efforts. Further studies beyond the scope of this paper will address discovery of compounds specifically targeting and reducing expression of hnRNPH2 via a HTS effort and subsequent cascade of assays.
Conclusion
As a result of the studies described herein, we developed novel High Content Assay for down regulators of hnNRPH2 amenable to the high throughput screening. A screen of FDA drug library identified two compounds that down regulate hnNRPH2 via dissimilar mechanisms and inhibit viability of malignant melanoma cells. Further studies are needed to dissect the mechanistic differences of hit compounds’ effects. In summary, our novel assay is suitable platform for large scale screening for novel down regulators of hnNRPH2. To our knowledge, this is a first HTS/HCS assay that allows monitoring of hnNRPH2 protein expression in live cells.
Authors thank Drs. Brent Samson and Steven T. Frank (ThermoFisher) for help with CX7 method development. DM envisioned and designed the study, performed HCS assay development and pilot screen experiments, analyzed all experimental data, and wrote the manuscript. JD developed WM266-4-H2GFP cell line, SB and PS performed hit validation western blot experiments. SR, TPS and LSD performed 1,536 well plate experiments. All authors have given approval to the final version of the manuscript.
Funding
This work was supported by the National Institutes of Health (AR066676 and CA249788 to DM).
The authors declare that no conflicts of interest exist.
| 1 Mei XL, Wei FL, Jia LL, Ji YZ: An alternative pathway for cellular protection in BRAF inhibitor resistance in aggressive melanoma type skin cancer. Chem Biol Interact 2020;323:109061. https://doi.org/10.1016/j.cbi.2020.109061 |
||||
| 2 Hartman ML, Sztiller-Sikorska M, Gajos-Michniewicz A, Czyz M: Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity. Cells 2020;9:142. https://doi.org/10.3390/cells9010142 |
||||
| 3 Palrasu M, Knapinska AM, Diez J, Smith L, LaVoi T, Giulianotti M, Houghten RA, Fields GB, Minond D: A Novel Probe for Spliceosomal Proteins that Induces Autophagy and Death of Melanoma Cells Reveals New Targets for Melanoma Drug Discovery. Cell Physiol Biochem 2019;53:656-686. https://doi.org/10.33594/000000164 |
||||
| 4 Konig M, Siegmund D, Raszeja LJ, Prokop A, Metzler-Nolte N: Resistance-breaking profiling and gene expression analysis on an organometallic Re(I)-phenanthridine complex reveal parallel activation of two apoptotic pathways. Medchemcomm 2018;9:173-180. https://doi.org/10.1039/C7MD00545H |
||||
| 5 Gowda R, Sharma A, Robertson GP: Synergistic inhibitory effects of Celecoxib and Plumbagin on melanoma tumor growth. Cancer Lett 2017;385:243-250. https://doi.org/10.1016/j.canlet.2016.10.016 |
||||
| 6 Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, Zhang D, Guha M, Xiao M, Randell S, Sproesser K, Xu W, Liu J, et al.: Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest 2016;126:1834-1856. https://doi.org/10.1172/JCI82661 |
||||
| 7 Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye WC, Zhang DM, Chen ZS: Autophagy and multidrug resistance in cancer. Chin J Cancer 2017;36:52. https://doi.org/10.1186/s40880-017-0219-2 |
||||
| 8 Martin S, Dudek-Peric AM, Garg AD, Roose H, Demirsoy S, Van Eygen S, Mertens F, Vangheluwe P, Vankelecom H, Agostinis P: An autophagy-driven pathway of ATP secretion supports the aggressive phenotype of BRAF(V600E) inhibitor-resistant metastatic melanoma cells. Autophagy 2017;13:1512-1527. https://doi.org/10.1080/15548627.2017.1332550 |
||||
| 9 Mowers EE, Sharifi MN, Macleod KF: Autophagy in cancer metastasis. Oncogene 2017;36:1619-1630. https://doi.org/10.1038/onc.2016.333 |
||||
| 10 Li CJ, Liao WT, Wu MY, Chu PY: New Insights into the Role of Autophagy in Tumor Immune Microenvironment. Int J Mol Sci 2017;18:1566. https://doi.org/10.3390/ijms18071566 |
||||
| 11 Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H: Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013;4:e838. https://doi.org/10.1038/cddis.2013.350 |
||||
| 12 Baillargeon P, Fernandez-Vega V, Sridharan BP, Brown S, Griffin PR, Rosen H, Cravatt B, Scampavia L, Spicer TP: The Scripps Molecular Screening Center and Translational Research Institute. SLAS Discov 2019;24:386-397. https://doi.org/10.1177/2472555218820809 |
||||
| 13 Lee KH, Lawley TJ, Xu YL, Swerlick RA: VCAM-1-, ELAM-1-, and ICAM-1-independent adhesion of melanoma cells to cultured human dermal microvascular endothelial cells. J Invest Dermatol 1992;98:79-85. https://doi.org/10.1111/1523-1747.ep12495643 |
||||
| 14 Ghosh D, Koch U, Hadian K, Sattler M, Tetko IV: Luciferase Advisor: High-Accuracy Model To Flag False Positive Hits in Luciferase HTS Assays. J Chem Inf Model 2018;58:933-942. https://doi.org/10.1021/acs.jcim.7b00574 |
||||
| 15 Thorne N, Auld DS, Inglese J: Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr Opin Chem Biol 2010;14:315-324. https://doi.org/10.1016/j.cbpa.2010.03.020 |
||||
| 16 Dantas RF, Evangelista TCS, Neves BJ, Senger MR, Andrade CH, Ferreira SB, Silva-Junior FP: Dealing with frequent hitters in drug discovery: a multidisciplinary view on the issue of filtering compounds on biological screenings. Expert Opin Drug Discov 2019:1-14. https://doi.org/10.1080/17460441.2019.1654453 |
||||
| 17 Fitzsimmons SA, Workman P, Grever M, Paull K, Camalier R, Lewis AD: Reductase enzyme expression across the National Cancer Institute Tumor cell line panel: correlation with sensitivity to mitomycin C and EO9. J Natl Cancer Inst 1996;88:259-269. https://doi.org/10.1093/jnci/88.5.259 |
||||
| 18 Snodgrass RG, Collier AC, Coon AE, Pritsos CA: Mitomycin C inhibits ribosomal RNA: a novel cytotoxic mechanism for bioreductive drugs. J Biol Chem 2010;285:19068-19075. https://doi.org/10.1074/jbc.M109.040477 |
||||
| 19 Filippov V, Filippova M, Duerksen-Hughes PJ: The early response to DNA damage can lead to activation of alternative splicing activity resulting in CD44 splice pattern changes. Cancer Res 2007;67:7621-7630. https://doi.org/10.1158/0008-5472.CAN-07-0145 |
||||
| 20 Uren PJ, Bahrami-Samani E, de Araujo PR, Vogel C, Qiao M, Burns SC, Smith AD, Penalva LO: High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol 2016;13:400-411. https://doi.org/10.1080/15476286.2015.1138030 |
||||
| 21 Lim LA, Madigan MC, Conway RM: Conjunctival melanoma: a review of conceptual and treatment advances. Clin Ophthalmol 2013;6:521-531. https://doi.org/10.2147/OPTH.S38415 |
||||
| 22 Patel C, Yan GX, Kowey PR: Dronedarone. Circulation 2009;120:636-644. https://doi.org/10.1161/CIRCULATIONAHA.109.858027 |
||||
| 23 Belur Nagaraj A, Joseph P, Kovalenko O, Wang Q, Xu R, DiFeo A: Evaluating class III antiarrhythmic agents as novel MYC targeting drugs in ovarian cancer. Gynecol Oncol 2018;151:525-532. https://doi.org/10.1016/j.ygyno.2018.09.019 |
||||
| 24 Elliott MJ, Jerzak KJ, Cockburn JG, Safikhani Z, Gwynne WD, Hassell JA, Bane A, Silvester J, Thu KL, Haibe-Kains B, Mak TW, Cescon DW: The Antiarrhythmic Drug, Dronedarone, Demonstrates Cytotoxic Effects in Breast Cancer Independent of Thyroid Hormone Receptor Alpha 1 (THRalpha1) Antagonism. Sci Rep 2018;8:16562. https://doi.org/10.1038/s41598-018-34348-0 |
||||
| 25 Shaughnessy M, Lamuraglia G, Klebanov N, Ji Z, Rajadurai A, Kumar R, Flaherty K, Tsao H: Selective uveal melanoma inhibition with calcium channel blockade. Int J Oncol 2019;55:1090-1096. https://doi.org/10.3892/ijo.2019.4873 |
||||