×
![]()
Corresponding Author: Albrecht Schwab
Institut für Physiologie II, Westfälische Wilhelms-Universität Münster
Robert-Koch-Str. 27b, 48149 Münster (Germany)
Tel. 0049251 8355329 , E-Mail aschwab@uni-muenster.de
Targeting Kca3.1 Channels in Cancer
Luca Matteo Todescaa Sarah Maskrib Kathrin Brömmelc Insa Thaleb Bernhard Wünschb Oliver Kochb Albrecht Schwaba
aInstitut für Phyisologie II, Westfälische Wilhelms-Universität Münster, Münster, Germany, bInstitute for Pharmaceutical and Medicinal Chemistry, Westfälische Wilhelms-Universität Münster, Münster, Germany, cMedizinische Klinik A, Westfälische Wilhelms-Universität Münster, Münster, Germany
Introduction
The Kca3.1 channel (also known as IKCa, SK4 channel), encoded by the KCNN4 gene, belongs to the family of Ca2+-activated K+ channels, that constitute a heterogeneous family of K+ channels with different biophysical and pharmacological properties, gated by Ca2+ [1]. It is a homotetrameric transmembrane protein whose subunits have six transmembrane domains, cytosolic N- and C-termini and a pore region between the 5th and 6th transmembrane domains [2]. Its activation by intracellular Ca2+ (100-500nM) is mediated by calmodulin (CaM) binding to its domain (CaMBD) which is located in the membrane-proximal region of the C-terminus of the channel protein [3, 4]. In non-excitable cells Kca3.1 -mediated K+ efflux results in a hyperpolarization of the cell membrane potential, which – in a positive feedback – can enhance the electrical driving force for Ca2+ ions to further enter the cell. The hyperpolarization and the increase of the intracellular Ca2+ concentration could be seen as an indirect read-out of Kca3.1 channel activity [5, 6].
Kca3.1 gating is also influenced by other molecular factors and interactions with cell signaling pathways. One example is the regulation by cAMP-dependent protein kinase (PKA) [7]. Phosphorylation of S334 reduces CaM binding to the channel and consequently the Kca3.1 mediated current. Physiologically, this may be triggered by activation of the adenosine A2a receptor. This decreases the open probability in wild-type but not S334A mutant Kca3.1 channels [8]. Studies on heterologously expressed canine Kca3.1 (cIK1) channels cloned from MDCK cells also showed an impact of protein kinase C (PKC) on the channel activity. Stimulation of PKC leads to an activation of the channel while mutating the PKC phosphorylation sites (T101, S178, T329) alters the regulation of the channel by PKC [9]. The channel is also activated by a nucleoside diphosphate Kinase-B (NDPK-B)-dependent phosphorylation of the histidine residue H358 [10].
Kca3.1 channels are widely expressed in many organs. Their expression has been demonstrated in human enteric, sensory and sympathetic neurons. In the CNS, however, they are mainly localized in microglia in which they are involved in their activation. Inhibition of Kca3.1 channels elicits neuroprotective effects [11]. Kca3.1 channels are also expressed in secretory epithelial cells of the gastro-intestinal tract as well as in exocrine glands such as the pancreas [12, 13]. In addition, the channel plays a role in cell migration, proliferation, activation and cytokine release of essentially all blood cells [14, 15]. According to their wide spread distribution, Kca3.1 channels play a role in many (inflammatory) pathologies, such as neurological disorders (e.g. ischemic stroke and multiple sclerosis), but also in vascular diseases such as atherosclerosis and restenosis [16-18].
Moreover, Kca3.1 channels have been studied intensively in the context of cancer, which is in the focus of this review. Their elevated expression in cancer cells has been linked to cancer progression and can be a valid marker and prognostic tool. Altered functions of the Kca3.1 channel are also proposed to promote oncogenic functions, suggesting the channel as a possible therapeutic target [19]. This aspect will be discussed in more detail in the following sections.
Kca3.1 channel functions in cancer
Kca3.1 expression in cancer
Based on qRT-PCR results Kca3.1 mRNA expression is 2-fold higher in clear cell renal cell carcinoma (ccRCC) than in the healthy tissue. The Kca3.1 high subgroup has a reduced progression-free survival [20]. The same findings were also demonstrated in papillary thyroid cancer where Kca3.1 mRNA levels are higher than in normal thyroid tissues. The high expression is associated with a larger tumor size, lymph node metastasis and advanced disease stage [21]. In human prostate cancer Kca3.1 channels are expressed grade-dependently [22]. Our group investigated why Kca3.1 channels are overexpressed in non-small cell lung cancer. Like in the above-mentioned cancer entities a Kca3.1 high phenotype also predicts a poor survival of non-small cell lung cancer patients. A genome-wide DNA methylation analysis revealed that Kca3.1 overexpression is due to hypomethylation of its promoter [5]. In cervical cancer tissue the elevated Kca3.1 expression (also correlated to the histological grade) has been proposed to result from a positive feedback. Increased Kca3.1 channel activity hyperpolarizes the cell membrane potential and leads to increased Ca2+ influx, which in turn activates the transcription factor AP-1 and post-translational histone acetylation to further drive the expression of the channel [23]. The correlation between histone acetylation and Kca3.1 channel expression was also shown in breast and prostate cancer cells lines (YMB-1 and PC-3, respectively). Accordingly, vorinostat, a histone deacetylase inhibitor, downregulates Kca3.1 transcription [24].
Altered mRNA expression of Kca3.1 channels in cancer has also been investigated with tissue microarrays [25]. Thus, characterization of a set of 10 K+ channels including Kca3.1 in a lung cancer microarray has shown its up-regulation and linkage to tumor aggressiveness. In addition to Kca3.1 channels, three other K+ channels, namely K2P1.1, K2P12.1, K2P5.1 and an accessory subunit of KCa1.1 channels, KCNMB3, are upregulated, too [26]. Likewise, the expression of Kca3.1 channels is increased in tumor cells isolated from micro-dissected samples of pancreatic ductal adenocarcinoma (PDAC) and associated with unfavorable overall survival [27, 28]. Finally, in serous ovarian carcinoma patients the high expression of Kca3.1 and S100A14 in micorarrays also has predictive value since it is associated with a high incidence of recurrence [29].
Role of Kca3.1 channels in cancer
Kca3.1 channels are not only a marker and a valid prognostic tool for cancer patients. They also have an important share in driving cancer progression because their function is required for many traits of the aggressive cancer cell behavior such as migration, invasion and metastasis, as well as proliferation and therapy resistance.
Kca3.1 is a promoter of cell cycle progression and cell proliferation. Its indirect role in driving Ca2+ influx into the cell allows the passing of G0/G1 and G1/S checkpoints which are regulated by the intracellular Ca2+ concentration [30]. Thus, blocking Kca3.1 channels in the breast cancer cell line MCF-7 with clotrimazole depolarizes the cell membrane potential, decreases the intracellular Ca2+ concentration and inhibits cell proliferation. The cells accumulate in the G1 phase and the number of cells in the S phase of the cell cycle is reduced [31]. Likewise, treating murine breast cancer cells (MMTV-PyMT) with the Kca3.1 blocker TRAM-34 inhibits cell cycle progression. The specificity of the pharmacological blockade of Kca3.1 channels was underpinned by the observation that the channel blockers mimic Kca3.1-/- breast cancer cell behavior [32]. Similar results were obtained in chronic lymphocytic leukemia (CCL) [33] and glioblastoma cells [34]. The latter study showed that the G2/M check-point is also regulated by Kca3.1 function via the activity of cdc25C phosphatase: activation of the channel with SKA-31 increases cdc2 phosphorylation [34].
These in vitro results are confirmed in in vivo studies. The volume of the cancer mass, resulting from the injection of human endometrial cancer cells (HEC-1-A) into nude mice, is decreased when mice are treated with TRAM-34 or clotrimazole [35]. The same has been shown in xenografted mice bearing intrahepatic cholangiocarcinoma Huh28 tumor cells or non-small cell lung cancer cells (A549-3R) that were treated with senicapoc [5, 36]. PET imaging also confirmed the inhibition of proliferation of lung cancer cells in vivo [5].
Kca3.1 channels seem to cooperate with different Ca2+ permeable channels. In the PDAC cell lines BxPC-3 and MiaPaCa-2 Kca3.1-regulated Ca2+ influx appears to occur via CRAC channels [37]. In prostate cancer cells the same mechanism is sustained by TRPV6 channels. Down-regulation of TRPV6 channels with siRNA decreases Ca2+ influx in response to Kca3.1 activation [38]. TRPC1 and Kca3.1 cooperate within lipid raft micro-domains in human breast cancer cells (MCF-7) where knockdown of TRPC1 suppresses Ca2+ influx induced by Kca3.1 activation with 1-EBIO [39]. In this context TRPC3 is yet another cooperation partner of Kca3.1 channels in pancreatic stellate cells [40].
The link between Kca3.1 expression and patient survival suggests that Kca3.1 channels are contributing to steps of the metastatic cascade, because almost all cancer patients die of the sequelae of metastasis. Several mechanisms by which Kca3.1 channels promote metastasis have been identified. Cell migration is one of the key processes of the metastatic cascade. Notably, Kca3.1 is one of the very first channels for which a role in cell migration was shown in transformed renal epithelial cells [41]. In the meantime, this initial observation has been reproduced and confirmed in many cancer cells. Examples include pancreatic, hepatocellular or non-small cell lung cancer cells to name only a few [5, 42-44]. Moreover, the inhibition of the channel with TRAM-34 also reduces glioblastoma infiltration in vivo. Brain slices from SCID mice in which GL-15 glioblastoma cells where injected into the brain, show a reduction of the tumor-infiltrated area in TRAM-34 treated mice [45]. One of the common signaling events by which Kca3.1 channels affect cell migration is the modulation of the intracellular Ca2+ concentration. In addition, they have an impact on the (local) regulation of the cell volume of migrating tumor cells [15].
Kca3.11 channels also regulate cancer cell apoptosis. This function has been linked to their expression in the inner mitochondrial membrane of the cell [46]. The localization of the channel in the inner mitochondrial membrane has been demonstrated in colon carcinoma and human cervix adenocarcinoma cells (HeLa) cells by means of Western blot and patch–clamp recordings [47, 48]. In melanoma cells (A-375) Kca3.1 channels play a key role in sensitizing them to TRAIL-induced apoptosis. Treating the cells both with TRAIL and TRAM-34 stimulates mitochondrial release of cytochrome c and starts the caspase cascade. TRAM-34 induces a hyperpolarization of the mitochondrial membrane potential and enhanced Bax translocation to the inner mitochondrial membrane [49]. Induction of the intrinsic apoptotic pathway in D54-MG glioma cells with staurosporine triggers a K+ efflux through the membrane via Kca3.1 channels which is followed by caspase 3 activation and apoptotic volume decrease (AVD) [50]. Finally, blocking Kca3.1 in hepatocellular carcinoma HepG2 cells induces apoptosis due to an increase of the intracellular ROS production and translocation of p53 protein from the cytoplasm into the nuclei [43].
Therapy resistance is a serious clinical problem in oncology which has not yet been overcome and which requires the development of new concepts. There is increasing evidence that ion channel modulation can help to overcome therapy resistance [51]. This also applies to Kca3.1 channels. In human epidermoid cancer cells resistant to cisplatin, Kca3.1 plays a crucial role in regulating sensitivity and resistance to cisplatin due to a control of proliferation and regulation of cell volume. Thus, Kca3.1 activation with 1-EBIO can promote apoptosis in cisplatin-resistant cells [52]. The same effect is observed in colorectal cell lines resistant to cisplatin (HCT-116 and HCT-8); application of Kca3.1 activator SKA-31 and inhibition of KV11.1 by E4031, have a synergistic effect with cisplatin and inhibit proliferation and activate apoptosis. Kca3.1 activators promote cisplatin uptake, and this effect further increases when Kca3.1 expression rises upon KV11.1 inhibition [53]. This is yet another example showing that Kca3.1 channels do not only act by themselves, but are rather as part of a network of functionally interacting channel proteins. The effect of Kca3.1 channels on drug resistance appears to depend both on the drug and on the tumor. Malignant glioma cells (GL261) are sensitized to temozolomide upon Kca3.1 inhibition. The combined treatment of TRAM-34 and temozolomide reduces cancer cell infiltration and migration more efficiently than the single drug [54]. Furthermore, in breast cancer overexpression of Kca3.1 enhances resistance to chemotherapeutic antimetabolites like gemcitabine. It does so by upregulating BCL2-related protein A1 (BCL2A1) which then suppresses apoptosis via RAS-MAPK and PI3K-AKT signaling. BCL2A1 is a member of the anti-apoptotic BCL-2 family that has been shown to confer resistance to anticancer drugs [55].
The role ofKca3.1 channels in the cancer microenvironment
So far, we have discussed the role of Kca3.1 channels in tumor cells. However, tumor cells do not act by themselves but are supported by cells of the tumor microenvironment located in the tumor stroma [56]. There is increasing evidence that ion channels expressed in tumor stroma cells such as immune cells, cancer-associated fibroblasts and endothelial cells play a crucial role in microenvironment remodeling and contribute to tumor progression [57, 58]. This also applies to Kca3.1 channels. In this context, immune cells belong to the best studied cells of the tumor microenvironment: The high K+ concentration found in the cancer microenvironment leads to an increased intracellular K+ concentration that can suppress T cell function by impairing Akt and PI3K pathways. Overexpression of KV1.3 and Kca3.1 in T cells enhance K+ release and thereby restore T cell function and decrease cancer growth [59, 60]. Kca3.1 channels also regulate the response of Natural Killer (NK) cells against tumors, in particular in adherent NK (A-NK) cells where Kca3.1 channels are up-regulated. Blocking the channel with TRAM-34 increases the degranulation, cytotoxicity and proliferation of A-NK cells. Moreover, TRAM-34 increases the ability of A-NK cells to reduce tumor growth [61]. In colorectal cancer, tumor-associated macrophages (TAMs) promote cancer progression through the release of cytokines such as IL-6 and IL-8. These effects can be attenuated by blocking Kca3.1 channels with TRAM-34, confirming the role of the channel in cytokine secretion and cancer invasion [62]. These examples clearly show that the final result of blocking Kca3.1 channels on tumor immunity is difficult to predict and will most likely depend on the individual tumor type. We refer to a recent review on the role of ion channels in PDAC where we extensively discuss the exciting question of ion channel function in tumor immunity [63].
In PDAC pancreatic stellate cells play an important role. Once activated they favor proliferation and metastasis of pancreatic cancer cells [64]. Kca3.1 is functionally expressed in pancreatic stellate cells and TRAM-34 treatment or Kca3.1 knockout inhibits their migration and chemotaxis. This is due to a decrease of the intracellular Ca2+ concentration which in turn leads to diminished calpain activity [39].
So far, there is no direct evidence for Kca3.1-dependent tumor angiogenesis. However, it is well known that the channel promotes non-cancerous angiogenesis. When human umbilical vein endothelial cells (HUVECs) and human microvascular endothelial cells 1 (HMEC-1) are stimulated with angiogenetic factors such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF), Kca3.1 channels are upregulated and their blockade suppresses angiogenesis in vivo [65]. Likewise, proliferation of vascular smooth muscle cells depends on Kca3.1 channel activity [66]. Therefore, it is plausible to assume that Kca3.1 inhibition will also impair tumour angiogenesis.
There is one aspect of the tumor microenvironment which has not yet been properly addressed in the context of ion channel function (see [67] for review). The microenvironment of solid tumors is usually quite acidic because of a metabolic shift of cancer cells that elicit an accumulation of metabolic acids in the tissue. Thus, the intracellular pH of the cells within the tumor microenvironment is also more acidic than in the healthy tissue. Many ion channels including Kca3.1 are pH-sensitive. In C6 rat glioma cells an intracellular acidification in the range of pHi 6.4–5.4 reduces channel open probability [68]. One could speculate in light of these findings that the upregulation of Kca3.1 channels in solid tumors is a compensatory mechanism in order to overcome the effect of reduced channel activity in an acidic environment. Hypoxia is another characteristic feature of the tumor microenvironment. It has been demonstrated in melanoma cell lines that hypoxia and hypoxia mimetics cause an upregulation of Kca3.1 channels [69, 70].
Imaging Kca3.1 channels in cancer cells
Several pharmacological modulators have been developed to either activate or inhibit Kca3.1 channel activity. Recent reviews give an overview about these compounds which are employed to modulate the function of the channel and thereby achieve a therapeutic effect when used clinically [71]. Channel activity itself can only be imaged indirectly by assessing the impact of the channel on the cell membrane potential or on the intracellular Ca2+ concentration. In addition, small molecules and channel targeting antibodies can be employed for imaging of the presence of Kca3.1 channels both in vitro and in vivo. Since Kca3.1 channel expression has predictive potential with respect to prognosis and patient survival in different tumor entities, imaging probes targeting the expression of this ion channel could serve as important diagnostic tools [72, 73].
Biological structures can be labeled, among others, by positron emission tomography (PET) tracers, antibodies and fluorescent dyes. A broad range of PET tracers has been developed to target proteins offering a distinct binding site. Thus, PET tracers are available to image ligand-gated ion channels such as nicotinic acetylcholine receptor subtypes [74] and NMDA receptors. Addressing specifically the ifenprodil binding site allows the selective imaging of NMDA receptors containing the GluN2B subunit [75-78]. However, PET tracers for imaging of voltage- or Ca2+-activated ion channels such as the Kca3.1 channel have not been developed so far.
Due to their high specificity, antibodies can be employed to image selectively almost all proteins. For example, the voltage-gated K+ channel KV11.1 (hERG1), which is aberrantly expressed in human tumors, can be visualized using an anti-KV11.1 monoclonal antibody. The expression of the KV11.1 channel in primary pancreatic ductal adenocarcinoma cells (PDAC) was investigated with this antibody in vitro and in vivo [79]. The fluorescent dye-coupled antibody mAb62-Cy5.5 was used to label the KV10.1 voltage-gated K+ channel of tumors in vitro and in vivo [80].
Antibody-based indirect immunofluorescence is one possibility to image Kca3.1 channels in vitro. Most antibodies available so far (e. g. AV35098 from Sigma-Aldrich) require fixation and permeabilization of (cancer) cells [5]. The above mentioned anti-Kca3.1 antibody binds from the cytosolic side to closedKca3.1 channels, which is the prevalent channel conformation in resting, non-activated cells. Imaging of Kca3.1 channels using the antibody-based indirect immunofluorescence is expensive and time-consuming due to long incubation times. Moreover, usually immobilization and permeabilization of the tumor cells are required for imaging with antibodies.
Alternatively to large and expensive antibodies, various fluorescently labeled small molecule probes have been developed to image ion channels, e.g. NMDA receptors [81, 82] and KV11.1 channels [83]. Some important features have to be considered when employing small molecule-based imaging probes: The pharmacophoric structural element interacting with the target channel must bind tightly to its binding site at the channel. Moreover, additional functional groups have to be present allowing chemical modification and coupling of the fluorescent dye via a linker. In particular, the linker length has to be carefully considered, since it can have an impact on the activity of the probe. Using poly(oxyethylene) linkers with different length will reduce the lipophilicity of the complete molecule. Additionally, the fluorescent dye must have a suitable excitation and emission profile for the available microscope recording system and a high intensity of its emitted light (high quantum yield). Furthermore, the excitation and emission wavelengths of the dye have to be outside of the region, where cells exhibit a high degree of autofluorescence and light scattering [82].
An interesting strategy for visualization of ion channels is the use of dyes with a turn-on mechanism which will lead to a reduction of background signals. Such a probe has been developed for imaging of KV11.1 channels. The probe contains the 2,7-dichlorofluorescein fluorophore, which is quenched intramolecularly by the basic amino moiety (electron donor) within the incorporated piperazine ring. Once the fluorescent probe binds to the target, the conformation changes, the fluorophore is no longer quenched and a fluorescence signal appears [82]. Based on this concept, other off-on imaging probes have been developed, which are partly based on solvatochromism or molecular rotation [84].
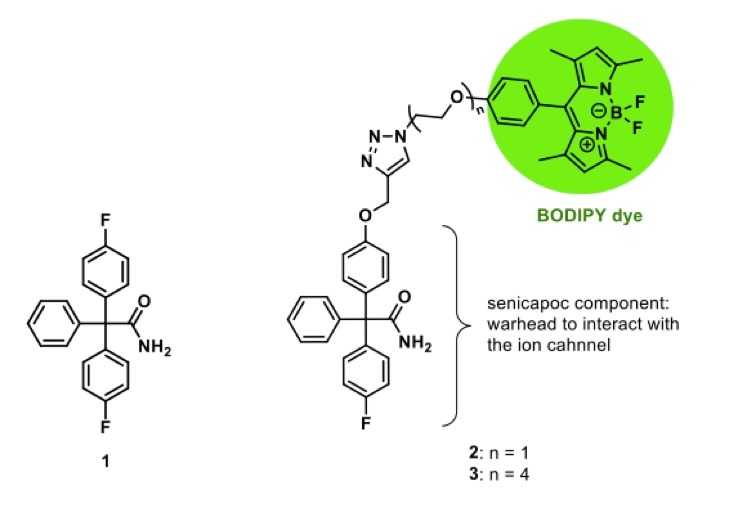
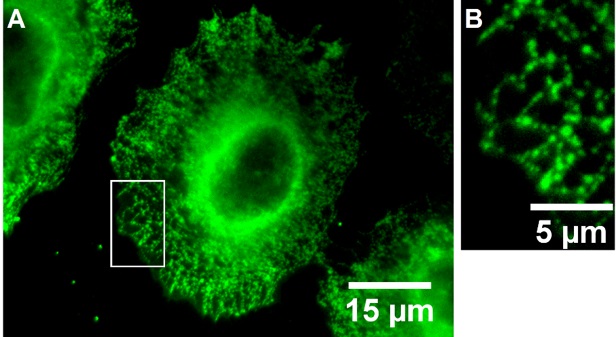
Fluorescently labeled small molecules targeting the Kca3.1channel can also be used for imaging the ion channel [74]. For this purpose, senicapoc (1), a highly potent and selective inhibitor of Kca3.1 channels, was connected with various BODIPY dyes via linkers of different length (Fig. 1). Both dimethylpyrrole-based BODIPY senicapoc conjugates 2 and 3 allow the visualization of Kca3.1 channels after an incubation period of only 10 min. Staining of Kca3.1 channels with the fluorescently labeled senicapoc-derivatives and the indirect immunofluorescence lead to the same typical punctate pattern [85, 86] (Fig. 2). The simple, fast and efficient staining protocol with small molecules can be performed with fixed cells without permeabilization. Moreover, the BODIPY-labeled senicapoc-derivative 2 with an ethoxy linker showed promising results in living cancer cells as well. It was postulated that BODIPY-labeled senicapoc derivatives 2 and 3 and anti-Kca3.1 antibodies label different populations of the Kca3.1 channel, which prevents co-staining with both methods. The anti-Kca3.1 antibody (Sigma #AV35098) labels the closed conformation of the channel, whereas the fluorescently labeled senicapoc derivatives 2 and 3 have to enter the open channel and thus bind to open Kca3.1 channels [87] (see below for a more detailed discussion).
Altogether, the BODIPY-labeled senicapoc derivatives 2 and 3 could serve as interesting new imaging tools for the diagnosis and/or monitoring of cancer patients, since they allow high throughput screening by fast and selective visualization of Kca3.1 channels.
This work was supported by the Deutsche Forschungsgemeinschaft (GRK 2515/1, Chembion).
The authors declare no conflict of interest.
| 1 Vergara C, Latorre R, Marrion N, Adelman J: Calcium-activated potassium channel. Curr Opin Neurobiol 1998;8:321-329. https://doi.org/10.1016/S0959-4388(98)80056-1 |
||||
| 2 Sforna L, Megaro A, Pessia M, Franciolini F, Catacuzzeno L, Structure: Gating and basic functions of the Ca2+-activated K channel of intermediate conductance. Curr Neuropharmacol 2018;16:608-617. https://doi.org/10.2174/1570159X15666170830122402 |
||||
| 3 Fanger CM, Ghanshani S, Logsdon NJ, Rauer H, Kalman K, Zhou J, Beckingham K, Chandy KG, Cahalan MD, Aiyar J: Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J Biol Chem 1999;274:5746-5754- https://doi.org/10.1074/jbc.274.9.5746 |
||||
| 4 Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP: Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol 2004;554:255-261. https://doi.org/10.1113/jphysiol.2003.049072 |
||||
| 5 Bulk E, Ay AS, Hammadi M, Ouadid-Ahidouch H, Schelhaas S, Hascher A, Rohde C, Thoennissen NH, Wiewrodt R, Schmidt E, Marra A, Hillejan L, Jacobs AH, Klein HU, Dugas M, Berdel WE, Müller-Tidow C, Schwab A: Epigenetic dysregulation of Kca3.1 channels induces poor prognosis in lung cancer. Int J Cancer 2015;137:1306-1317. https://doi.org/10.1002/ijc.29490 |
||||
| 6 Yi M, Yu P, Lu Q, Geller HM, Yu Z, Chen H: Kca3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer's disease. Mol Cell Neurosci 2016;76:21-32. https://doi.org/10.1016/j.mcn.2016.08.008 |
||||
| 7 Gerlach AC, Gangopadhyay NN, Devor DC: Kinase-dependent regulation of the intermediate conductance, calcium-dependent potassium channel, hIK1. J Biol Chem 2000;275:585-598. https://doi.org/10.1074/jbc.275.1.585 |
||||
| 8 Wong R, Schlichter LC: PKA reduces the rat and human Kca3.1 current, CaM binding, and Ca2+ signaling, which requires Ser332/334 in the CaM-binding C terminus. J Neurosci 2014;34:13371-13383. https://doi.org/10.1523/JNEUROSCI.1008-14.2014 |
||||
| 9 Wulf A, Schwab A: Regulation of a calcium-sensitive K+ channel (cIK1) by protein kinase C. J Membr Biol 2002;187:71-79. https://doi.org/10.1007/s00232-001-0149-3 |
||||
| 10 Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele DJ, Hubbard SR, Skolnik EY. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel Kca3.1. Elife 2016;5:e16093. https://doi.org/10.7554/eLife.16093 |
||||
| 11 Kshatri AS, Gonzalez-Hernandez A, Giraldez T: Physiological roles and therapeutic potential of Ca2+ activated potassium channels in the nervous system. Front Mol Neurosci 2018;11:258. https://doi.org/10.3389/fnmol.2018.00258 |
||||
| 12 Chen MX, Gorman SA, Benson B, Singh K, Hieble JP, Michel MC, Tate SN, Trezise DJ: Small and intermediate conductance Ca2+-activated K+ channels confer distinctive patterns of distribution in human tissues and differential cellular localisation in the colon and corpus cavernosum. Naunyn Schmiedebergs Arch Pharmacol 2004;369:602-615. https://doi.org/10.1007/s00210-004-0934-5 |
||||
| 13 Wulff H, Castle NA: Therapeutic potential of Kca3.1 blockers: recent advances and promising trends. Expert Rev Clin Pharmacol 2010;3:385-396. https://doi.org/10.1586/ecp.10.11 |
||||
| 14 Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG: Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem 2000;275:37137-37149. https://doi.org/10.1074/jbc.M003941200 |
||||
| 15 Schwab A, Fabian A, Hanley PJ, Stock C: Role of ion channels and transporters in cell migration. Physiol Rev 2012;92:1865-1913. https://doi.org/10.1152/physrev.00018.2011 |
||||
| 16 Chou CC, Lunn CA, Murgolo NJ. Kca3.1: target and marker for cancer, autoimmune disorder and vascular inflammation? Expert Rev Mol Diagn 2008;8:179-187. https://doi.org/10.1586/14737159.8.2.179 |
||||
| 17 Sugunan S, Nampoothiri SS, Garg T, Krishnamurthy RG: Role of Kca3.1 channels in CNS diseases: A concise review. CNS Neurol Disord Drug Targets 2016;15:1299-1305. https://doi.org/10.2174/1871527315666160822111913 |
||||
| 18 Köhler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, Grgic I, Kämpfe D, Si H, Wibawa J, Real R, Borner K, Brakemeier S, Orzechowski HD, Reusch HP, Paul M, Chandy KG, Hoyer J: Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation 2003;108:1119-1125. https://doi.org/10.1161/01.CIR.0000086464.04719.DD |
||||
| 19 Mohr CJ, Steudel FA, Gross D, Ruth P, Lo WY, Hoppe R, Schroth W, Brauch H, Huber SM, Lukowski R: Cancer-Associated Intermediate Conductance Ca2+-Activated K⁺ Channel Kca3.1. Cancers (Basel) 2019;11:109. https://doi.org/10.3390/cancers11010109 |
||||
| 20 Rabjerg M, Oliván-Viguera A, Hansen LK, Jensen L, Sevelsted-Møller L, Walter S, Jensen BL, Marcussen N, Köhler R: High expression of Kca3.1 in patients with clear cell renal carcinoma predicts high metastatic risk and poor survival. PLoS One 2015;10:e0122992. https://doi.org/10.1371/journal.pone.0122992 |
||||
| 21 Wen J, Lin B, Lin L, Chen Y, Wang O: KCNN4 is a diagnostic and prognostic biomarker that promotes papillary thyroid cancer progression. Aging 2020;12:16437-16456. https://doi.org/10.18632/aging.103710 |
||||
| 22 Ohya S, Kimura K, Niwa S, Ohno A, Kojima Y, Sasaki S, Kohri K, Imaizumi Y: Malignancy grade-dependent expression of K+-channel subtypes in human prostate cancer. J Pharmacol Sci 2009;109:148-151. https://doi.org/10.1254/jphs.08208SC |
||||
| 23 Liu L, Zhan P, Nie D, Fan L, Lin H, Gao L, Mao X: Intermediate-conductance-Ca2+-activated K channel IKCa1 Is upregulated and promotes cell proliferation in cervical cancer. Med Sci Monit Basic Res 2017;23:45-57. https://doi.org/10.12659/MSMBR.901462 |
||||
| 24 Ohya S, Kanatsuka S, Hatano N, Kito H, Matsui A, Fujimoto M, Matsuba S, Niwa S, Zhan P, Suzuki T, Muraki K: Downregulation of the Ca2+-activated K+ channel Kca3.1 by histone deacetylase inhibition in human breast cancer cells. Pharmacol Res Perspect 2016;4:e00228. https://doi.org/10.1002/prp2.228 |
||||
| 25 Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D: Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 2000;16:906-914. https://doi.org/10.1093/bioinformatics/16.10.906 |
||||
| 26 Ko EA, Kim YW, Lee D, Choi J, Kim S, Seo Y, Bang H, Kim JH, Ko JH: Expression of potassium channel genes predicts clinical outcome in lung cancer. Korean J Physiol Pharmacol 2019;23:529-537. https://doi.org/10.4196/kjpp.2019.23.6.529 |
||||
| 27 Zaccagnino A, Pilarsky C, Tawfik D, Sebens S, Trauzold A, Novak I, Schwab A, Kalthoff H: In silico analysis of the transportome in human pancreatic ductal adenocarcinoma. Eur J Biophys 2016;45:749-763. https://doi.org/10.1007/s00249-016-1171-9 |
||||
| 28 Jiang S, Zhu L, Yang J, Hu L, Gu J, Xing X, Sun Y, Zhang Z: Integrated expression profiling of potassium channels identifys KCNN4 as a prognostic biomarker of pancreatic cancer. Biochem Biophys Res Commun 2017;494:113-119. https://doi.org/10.1016/j.bbrc.2017.10.072 |
||||
| 29 Zhao H, Guo E, Hu T, Sun Q, Wu J, Lin X, Luo D, Sun C, Wang C, Zhou B, Li N, Xia M, Lu H, Meng L, Xu X, Hu J, Ma D, Chen G, Zhu T: KCNN4 and S100A14 act as predictors of recurrence in optimally debulked patients with serous ovarian cancer. Oncotarget 2016;7:43924-43938. https://doi.org/10.18632/oncotarget.9721 |
||||
| 30 Sundelacruz S, Levin M, Kaplan DL: Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev Rep 2009;5:231-246. https://doi.org/10.1007/s12015-009-9080-2 |
||||
| 31 Ouadid-Ahidouch H, Roudbaraki M, Delcourt P, Ahidouch A, Joury N, Prevarskaya N: Functional and molecular identification of intermediate-conductance Ca2+-activated K+ channels in breast cancer cells: association with cell cycle progression. Am J Physiol Cell Physiol 2004;287:C125-C134. https://doi.org/10.1152/ajpcell.00488.2003 |
||||
| 32 Steudel FA, Mohr CJ, Stegen B, Nguyen HY, Barnert A, Steinle M, Beer-Hammer S, Koch P, Lo WY, Schroth W, Hoppe R, Brauch H, Ruth P, Huber SM, Lukowski R: SK4 channels modulate Ca2+ signalling and cell cycle progression in murine breast cancer. Mol Oncol 2017;11:1172-1188. https://doi.org/10.1002/1878-0261.12087 |
||||
| 33 Grössinger EM, Weiss L, Zierler S, Rebhandl S, Krenn PW, Hinterseer E, Schmölzer J, Asslaber D, Hainzl S, Neureiter D, Egle A, Piñón-Hofbauer J, Hartmann TN, Greil R, Kerschbaum HH: Targeting proliferation of chronic lymphocytic leukemia (CLL) cells through Kca3.1 blockade. Leukemia 2014;28:954-958. https://doi.org/10.1038/leu.2014.37 |
||||
| 34 D'Alessandro G, Limatola C, Catalano M: Functional Roles of the Ca2+-activated K+ channel, Kca3.1 , in brain tumors. Curr Neuropharmacol 2018;16:636-643. https://doi.org/10.2174/0929867324666170713103621 |
||||
| 35 Wang ZH, Shen B, Yao H, Jia YC, Ren J, Feng YJ, Wang YZ: Blockage of intermediate-conductance-Ca2+-activated K+ channels inhibits progression of human endometrial cancer. Oncogene 2007;26:5107-5114. https://doi.org/10.1038/sj.onc.1210308 |
||||
| 36 Song P, Du Y, Song W, Chen H, Xuan Z, Zhao L, Chen J, Chen J, Guo D, Jin C, Zhao Y, Tuo B, Zheng S: Kca3.1 as an effective target for inhibition of growth and progression of intrahepatic cholangiocarcinoma. J Cancer. 2017;8:1568-1578. https://doi.org/10.7150/jca.18697 |
||||
| 37 Jäger H, Dreker T, Buck A, Giehl K, Gress T, Grissmer S: Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol Pharmacol 2004;65:630-638. https://doi.org/10.1124/mol.65.3.630 |
||||
| 38 Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackière F, Bidaux G, Urbain R, Gosset P, Delcourt P, Fleurisse L, Slomianny C, Dewailly E, Mauroy B, Bonnal JL, Skryma R, Prevarskaya N: Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 2009;28:1792-1806. https://doi.org/10.1038/onc.2009.25 |
||||
| 39 Faouzi M, Hague F, Geerts D, Ay AS, Potier-Cartereau M, Ahidouch A, Ouadid-Ahidouch H: Functional cooperation between Kca3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016;7:36419-36435. https://doi.org/10.18632/oncotarget.9261 |
||||
| 40 Storck H, Hild B, Schimmelpfennig S, Sargin S, Nielsen N, Zaccagnino A, Budde T, Novak I, Kalthoff H, Schwab A: Ion channels in control of pancreatic stellate cell migration. Oncotarget 2017;8:769-784. https://doi.org/10.18632/oncotarget.13647 |
||||
| 41 Schwab A, Wojnowski L, Gabriel K, Oberleithner H: Oscillating activity of a Ca2+-sensitive K+ channel - a prerequisite for migration of alkali-transformed Madin-Darby canine kidney (MDCK-F) cells. J Clin Invest 1994;93:1631-1636 https://doi.org/10.1172/JCI117144 |
||||
| 42 Bonito B, Sauter DR, Schwab A, Djamgoz MB, Novak I: Kca3.1 (IK) modulates pancreatic cancer cell migration, invasion and proliferation: anomalous effects on TRAM-34. Pflugers Arch 2016;468:1865-1875. https://doi.org/10.1007/s00424-016-1891-9 |
||||
| 43 Li QT, Feng YM, Ke ZH, Qiu MJ, He XX, Wang MM, Li YN, Xu J, Shi LL, Xiong ZF: KCNN4 promotes invasion and metastasis through the MAPK/ERK pathway in hepatocellular carcinoma. J Investig Med 2020;68:68-74. https://doi.org/10.1136/jim-2019-001073 |
||||
| 44 Liu Y, Zhao L, Ma W, Cao X, Chen H, Feng D, Liang J, Yin K, Jiang X: The Blockage of Kca3.1 Channel inhibited proliferation, migration and promoted apoptosis of human hepatocellular carcinoma cells. J Cancer 2015;6:643-651. https://doi.org/10.7150/jca.11913 |
||||
| 45 D'Alessandro G, Catalano M, Sciaccaluga M, Chece G, Cipriani R, Rosito M, Grimaldi A, Lauro C, Cantore G, Santoro A, Fioretti B, Franciolini F, Wulff H, Limatola C: Kca3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis 2013;4:e773. https://doi.org/10.1038/cddis.2013.279 |
||||
| 46 Szabo I, Zoratti M: Mitochondrial channels: ion fluxes and more. Physiol Rev 2014:94:519-608. https://doi.org/10.1152/physrev.00021.2013 |
||||
| 47 De Marchi U, Sassi N, Fioretti B, Catacuzzeno L, Cereghetti GM, Szabò I, Zoratti M: Intermediate conductance Ca2+-activated potassium channel (Kca3.1 ) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009;45:509-516. https://doi.org/10.1016/j.ceca.2009.03.014 |
||||
| 48 Sassi N, De Marchi U, Fioretti B, Biasutto L, Gulbins E, Franciolini F, Szabò I, Zoratti M: An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKca3.1. Biochim Biophys Acta 2010;1797:1260-1267. https://doi.org/10.1016/j.bbabio.2009.12.015 |
||||
| 49 Quast SA, Berger A, Buttstädt N, Friebel K, Schönherr R, Eberle J: General sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS One 2012;7:e39290. https://doi.org/10.1371/journal.pone.0039290 |
||||
| 50 McFerrin MB, Turner KL, Cuddapah VA, Sontheimer H: Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am J Physiol Cell Physiol 2012;303:C1070-C1078. https://doi.org/10.1152/ajpcell.00040.2012 |
||||
| 51 Kischel P, Girault A, Rodat-Despoix L, Chamlali M, Radoslavova S, Abou Daya H, Lefebvre T, Foulon A, Rybarczyk P, Hague F, Dhennin-Duthille I, Gautier M, Ouadid-Ahidouch H. Ion channels: New actors playing in chemotherapeutic resistance. Cancers (Basel) 2019;11:376. https://doi.org/10.3390/cancers11030376 |
||||
| 52 Lee EL, Hasegawa Y, Shimizu T, Okada Y: IK1 channel activity contributes to cisplatin sensitivity of human epidermoid cancer cells. Am J Physiol Cell Physiol 2008;294:C1398-C1406. https://doi.org/10.1152/ajpcell.00428.2007 |
||||
| 53 Pillozzi S, D'Amico M, Bartoli G, Gasparoli L, Petroni G, Crociani O, Marzo T, Guerriero A, Messori L, Severi M, Udisti R, Wulff H, Chandy KG, Becchetti A, Arcangeli A: The combined activation of Kca3.1 and inhibition of KV11.1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br J Cancer 2018;118:200-212. https://doi.org/10.1038/bjc.2017.392 |
||||
| 54 D'Alessandro G, Grimaldi A, Chece G, Porzia A, Esposito V, Santoro A, Salvati M, Mainiero F, Ragozzino D, Di Angelantonio S, Wulff H, Catalano M, Limatola C: Kca3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016;7:30781-30796. https://doi.org/10.18632/oncotarget.8761 |
||||
| 55 Lin P, Li J, Ye F, Fu W, Hu X, Shao Z, Song C: KCNN4 induces multiple chemoresistance in breast cancer by regulating BCL2A1. Am J Cancer Res 2020;10:3302-3315. | ||||
| 56 Falcone, Conciatori F, Bazzichetto C, Ferretti G, Cognetti F, Ciuffreda L, Milella M, Tumor microenvironment: Implications in melanoma resistance to targeted therapy and immunotherapy. Cancers (Basel) 2020;12:2870. https://doi.org/10.3390/cancers12102870 |
||||
| 57 Arcangeli A: Ion channels and transporters in cancer. 3. Ion channels in the tumor cell-microenvironment cross talk. Am J Physiol Cell Physiol 2011;301:C762-C771. https://doi.org/10.1152/ajpcell.00113.2011 |
||||
| 58 Arcangeli A, Crociani O, Bencini L: Interaction of tumour cells with their microenvironment: ion channels and cell adhesion molecules. A focus on pancreatic cancer. Philos Trans R Soc Lond B Biol Sci 2014;369:20130101. https://doi.org/10.1098/rstb.2013.0101 |
||||
| 59 Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, Guittard GC, Yu Z, Carbonaro V, Okkenhaug K, Schrump DS, Linehan WM, Roychoudhuri R, Restifo NP: Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016;537:539-543. https://doi.org/10.1038/nature19364 |
||||
| 60 Chandy KG, Norton RS: Immunology: Channelling potassium to fight cancer. Nature 2016;537:497-499. https://doi.org/10.1038/nature19467 |
||||
| 61 Koshy S, Wu D, Hu X, Tajhya RB, Huq R, Khan FS, Pennington MW, Wulff H, Yotnda P, Beeton C: Blocking Kca3.1 channels increases tumor cell killing by a subpopulation of human natural killer lymphocytes. PLoS One 2013;8:e76740. https://doi.org/10.1371/journal.pone.0076740 |
||||
| 62 Xu H, Lai W, Zhang Y, Liu L, Luo X, Zeng Y, Wu H, Lan Q, Chu Z: Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer 2014;14:330. https://doi.org/10.1186/1471-2407-14-330 |
||||
| 63 Hofschröer V, Najder K, Rugi M, Bouazzi R, Cozzolino M, Arcangeli A, Panyi G, Schwab A: Ion channels orchestrate pancreatic ductal adenocarcinoma progression and therapy. Front Pharmacol 2021;11:586599. https://doi.org/10.3389/fphar.2020.586599 |
||||
| 64 Hessmann E, Buchholz SM, Demir IE, Singh SK, Gress TM, Ellenrieder V, Neessee A: Microenvironmental determinants of pancreatic cancer. Physiol Rev 2020;100:1707-1751 https://doi.org/10.1152/physrev.00042.2019 |
||||
| 65 Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, Köhler R: Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol 2005;25:704-709. https://doi.org/10.1161/01.ATV.0000156399.12787.5c |
||||
| 66 Bi D, Toyama K, Lemaître V, Takai J, Fan F, Jenkins DP, Wulff H, Gutterman DD, Park F, Miura H: The intermediate conductance calcium-activated potassium channel Kca3.1 regulates vascular smooth muscle cell proliferation via controlling calcium-dependent signaling. J Biol Chem 2013;288:15843-15853. https://doi.org/10.1074/jbc.M112.427187 |
||||
| 67 Pethő Z, Najder K, Carvalho T, McMorrow R, Todesca LM, Rugi M, Bulk E, Chan A, Löwik CWGM, Reshkin SJ, Schwab A: pH-Channeling in Cancer: How pH-dependence of cation channels shapes cancer pathophysiology. Cancers (Basel) 2020;12:2484. https://doi.org/10.3390/cancers12092484 |
||||
| 68 Strupp M, Staub F, Graf PA: Ca2+- and pH-dependent K+ channel of rat C6 glioma cells and its possible role in acidosis-induced cell swelling. Glia 1993;9:136-145. https://doi.org/10.1002/glia.440090207 |
||||
| 69 Tajima N, Schönherr K, Niedling S, Kaatz M, Kanno H, Schönherr R, Heinemann SH: Ca2+-activated K+ channels in human melanoma cells are up-regulated by hypoxia involving hypoxia-inducible factor-1alpha and the von Hippel-Lindau protein. J Physiol 2006;571:349-359. https://doi.org/10.1113/jphysiol.2005.096818 |
||||
| 70 Girault A, Ahidouch A, Ouadid-Ahidouch H: Roles for Ca2+ and K+ channels in cancer cells exposed to the hypoxic tumour microenvironment. Biochim Biophys Acta Mol Cell Res 2020;1867:118644. https://doi.org/10.1016/j.bbamcr.2020.118644 |
||||
| 71 Brown BM, Shim H, Christophersen P, Wulff H: Pharmacology of small- and intermediate-conductance calcium-activated potassium channels. Annu Rev Pharmacol Toxicol 2020;60:219-240. https://doi.org/10.1146/annurev-pharmtox-010919-023420 |
||||
| 72 Haren N, Khorsi H, Faouzi M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H: Intermediate conductance Ca2+ activated K+ channels are expressed and functional in breast adenocarcinomas: correlation with tumour grade and metastasis status. Histol Histopathol 2010;25:1247-1255. | ||||
| 73 Ko JH, Ko EA, Gu W, Lim I, Bang H, Zhou T: Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Mol Cancer 2013;12:106. https://doi.org/10.1186/1476-4598-12-106 |
||||
| 74 Mo Y, Yin Y, Li Y: Neural nAChRs PET imaging probes. Nucl Med Commun 2014;35:135-143. https://doi.org/10.1097/MNM.0000000000000032 |
||||
| 75 Klein PJ, Christiaans JA, Metaxas A, Schuit RC, Lammertsma AA, van Berckel BN, Windhorst AD: Synthesis, structure activity relationship, radiolabeling and preclinical evaluation of high affinity ligands for the ion channel of the N-methyl-d-aspartate receptor as potential imaging probes for positron emission tomography. Bioorg Med Chem 2015;23:1189-206. https://doi.org/10.1016/j.bmc.2014.12.029 |
||||
| 76 Haider A, Herde AM, Krämer SD, Varisco J, Keller C, Frauenknecht K, Auberson YP, Temme L, Robaa D, Sippl W, Schibli R, Wünsch B, Mu L, Ametamey SM: Preclinical evaluation of benzazepine-based PET radioligands (R)- and (S)-11C-Me-NB1 reveals distinct enantiomeric binding patterns and a tightrope walk between GluN2B- and σ1-receptor-targeted PET imaging. J Nucl Med 2019;60:1167-1173. https://doi.org/10.2967/jnumed.118.221051 |
||||
| 77 Haider A, Iten I, Ahmed H, Müller Herder A, Gruber S, Krämer SD, Keller C, Schibli R, Wünsch B, Mu L, Ametamey SM: Identification and preclinical evaluation of a radiofluorinated benzazepine derivative for imaging the GluN2B subunit of the ionotropic NMDA receptor. J Nucl Med 2018;118:212134. https://doi.org/10.2967/jnumed.118.212134 |
||||
| 78 Ahmed H, Wallimann R, Haider A, Hosseini V, Gruber S, Robledo M, Nguyen TAN, Herde AM, Iten I, Keller C, Vogel V, Schibli R, Wuensch B, Mu L, Ametamey SM: Preclinical development of 18F-OF-NB1 for imaging GluN2B-Containing N-Methyl-D-aspartate receptors and its utility as a biomarker for amyotrophic lateral sclerosis. J Nucl Med 2020;120:246785. https://doi.org/10.2967/jnumed.120.246785 |
||||
| 79 Lastraioli E, Perrone G, Sette A, Fiore A, Crociani O, Manoli S, D'Amico M, Masselli M, Iorio J, Callea M, Borzomati D, Nappo G, Bartolozzi F, Santini D, Bencini L, Farsi M, Boni L, Di Costanzo F, Schwab A, Onetti Muda A, et al.: hERG1 channels drive tumour malignancy and may serve as prognostic factor in pancreatic ductal adenocarcinoma. Brit J Cancer 2015;112:1076-1087 https://doi.org/10.1038/bjc.2015.28 |
||||
| 80 Napp J, Pardo LA, Hartung F, Tietze LF, Stühmer W, Alves F: In vivo imaging of tumour xenografts with an antibody targeting the potassium channel Kv10.1. Eur Biophys J 2016;45:721-733. https://doi.org/10.1007/s00249-016-1152-z |
||||
| 81 Dhilly M, Becerril-Ortega J, Colloc'h N, MacKenzie ET, Barr L, Buisson A, Nicole O, Perrio C: Synthesis and in vitro characterisation of ifenprodil-based fluorescein conjugates as GluN1/GluN2B N-Methyl-D-aspartate receptor antagonists. Chembiochem 2013;14:759 - 769 https://doi.org/10.1002/cbic.201200675 |
||||
| 82 McCarron ST, Chambers JJ: Modular chemical probes for visualizing and tracking endogenous ion channels. Neuropharmacology 2015;98:41-47. https://doi.org/10.1016/j.neuropharm.2015.03.033 |
||||
| 83 Liu Z, Wang B, Ma Z, Zhou Y, Du L, Li M: Fluorogenic probe for the human Ether-a-Go-Go-Related Gene potassium channel imaging. Anal Chem 2015;87:2550-2554. https://doi.org/10.1021/ac504763b |
||||
| 84 Qiao Z, Zhou Q, Zhang H, Wei N, Zhang Y, Wang K: The visualization of hERG channels in living cells via a fluorescent probe regulated by the synergy between solvatochromism and molecular rotation based on simple targeting of the group 4-benzylaniline. Chem Commun (Camb) 2019;55:5515-5518. https://doi.org/10.1039/C9CC01735F |
||||
| 85 Brömmel K, Maskri S, Maisuls I, Konken CP, Rieke M, Pethő Z, Strassert CA, Koch O, Schwab A, Wünsch B: Synthesis of small-molecule fluorescent probes for the in vitro imaging of calcium-activated potassium channel Kca3.1. Angew Chem Int Ed 2020;132:8354-8361. https://doi.org/10.1002/ange.202001201 |
||||
| 86 Nechyporuk-Zloy V, Stock C, Schillers H, Oberleithner H, Schwab A: Single plasma membrane K+ channel detection by using dual-color quantum dot labeling. Am J Physiol Cell Physiol 2006;291:C266-C269. https://doi.org/10.1152/ajpcell.00633.2005 |
||||
| 87 Brömmel K, Maskri S, Bulk E, Pethő Z, Rieke M, Budde T, Koch O, Schwab A, Wünsch B: Co-staining of KCa 3.1 channels in NSCLC cells with a small-molecule fluorescent probe and antibody-based indirect immunofluorescence. ChemMedChem 2020;15:2462-2469. https://doi.org/10.1002/cmdc.202000652 |
||||
| 88 Lee CH and MacKinnon R: Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 2018;360:508-513. https://doi.org/10.1126/science.aas9466 |
||||
| 89 Garneau L, Klein H, Banderali U, Longpré-Lauzon A, Parent L, Sauvé R: Hydrophobic interactions as key determinants to the Kca3.1 channel closed configuration. An analysis of Kca3.1 mutants constitutively active in zero Ca2+. J Biol Chem 2009;284:389-403. https://doi.org/10.1074/jbc.M805700200 |
||||
| 90 Wulff H, Gutman GA, Cahalan MD, Chandy KG: Delineation of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel, IKCa1. J Biol Chem 2001;276:32040-32045. https://doi.org/10.1074/jbc.M105231200 |
||||
| 91 Nguyen HM, Singh V, Pressly B, Jenkins DP, Wulff H, Yarov-Yarovoy V: Structural insights into the atomistic mechanisms of action of small molecule inhibitors targeting the KCa3. 1 channel pore. Mol Pharmacol 2017;91:392-402. https://doi.org/10.1124/mol.116.108068 |
||||
| 92 Ataga KI, Staffa SJ, Brugnara C, Stocker JW: Hemoglobin response to senicapoc in patients with sickle cell disease: a re-analysis of the Phase III trial. Br J Haematol 2021;192:121-135. https://doi.org/10.1111/bjh.17345 |
||||
| 93 ClinicalTrials.gov: Senicapoc in Alzheimer's disease (Senicapoc), University of California, Davis (U.S.), Identifier: NCT04804241. URL: https://clinicaltrials.gov/ct2/show/NCT04804241?term=senicapoc&draw=1&rank=1. | ||||
| 94 Jin LW, Lucente JD, Nguyen HM, Singh V, Singh L, Chavez M, Bushong T, Wulff H, Maezawa I: Repurposing the Kca3.1 inhibitor senicapoc for Alzheimer's disease. Ann Clin Transl Neurol 2019;6:723-738. https://doi.org/10.1002/acn3.754 |
||||
| 95 Staal RGW, Weinstein JR, Nattini M, Cajina M, Chandresana G, Möller T: Senicapoc: Repurposing a drug to target microglia Kca3.1 in stroke. Neurochem Res 2017;42:2639-2645. https://doi.org/10.1007/s11064-017-2223-y |
||||