×
![]()
Corresponding Author: Guiscard Seebohm
Cellular Electrophysiology and Molecular Biology, Institute for Genetics of Heart Diseases (IfGH), University Hospital Münster (Germany)
E-Mail guiscard.seebohm@ukmuenster.de
Can Unlikely Neanderthal Chloride Channel CLC-2 Gene Variants Provide Insights in Modern Human Infertility?
Elena Jeworutzkia Frank Tüttelmannb Ina Rothenberga Michael Puschc Julian A. Schreibera,d Sabine Kliesche Bernhard Wünschd Nathalie Strutz-Seebohma Guiscard Seebohma
aCellular Electrophysiology and Molecular Biology, Institute for Genetics of Heart Diseases (IfGH), University Hospital Münster, Münster, Germany, bInstitute of Reproductive Genetics, University of Münster, Münster, Germany, cIstituto di Biofisica, Consiglio Nazionale delle Ricerche, Genova, Italy, dInstitute of Pharmaceutical and Medicinal Chemistry, University of Münster, Münster, Germany, eCentre of Reproductive Medicine and Andrology, Department of Clinical and Surgical Andrology, University Hospital Münster, Münster, Germany
Introduction
DNA comparisons between Neanderthals and AMH have indicated that Neanderthals mated with and share more genetic variants with non-Africans than with Africans [1]. At around 41,000 years ago, modern humans bearing the so-called Protoaurignacian culture spread into southern Europe leading to the demise of Neanderthals [2]. However, the reasons why Neanderthals were replaced by AMH are heavily discussed and include factors such as environmental changes leading to reduced genetic variation [3], changes in language [4], infectious diseases [5], behavior-cultural reasons [6], and anatomical and physiological cues [7, 8]. An additional driver for the Neanderthal demise may have been that they were simply outnumbered. Neanderthal populations were generally small in size and rather isolated, but shortly after the arrival of AMH, the human population increased tenfold for unknown reasons but AMH’s numerical supremacy may have driven Neanderthal replacement [9, 10].
Recently, the debate about what drove the extinction of Neanderthals has started to include physiological factors, such as metabolic peculiarities in Neanderthals’ lipid and glucose metabolism [7, 8]. Because such physiological issues are central players in current-day human health, they may have been even more critical for determining the survival of hominids like the small-group dwelling Neanderthals. Therefore, key elements of health in modern humans may have been relevant for Neanderthal fitness. We analyzed the rate of genetic variance in ion channel-encoding genes. Ion channels are such key players in human health and fitness, and certain human diseases called channelopathies arise from a series of ion channel genetic variants. Furthermore, and unsurprisingly, the majority of all current drugs on the market act directly or indirectly via ion channels. Therefore, ion channel gene variants represent perfect candidates for evolutionary drivers of fitness (or lack thereof).
Materials and Methods
Mutagenesis and Electrophysiology. Human CLCN2 was subcloned into pSGEM. hKv1.5-eGFP was obtained by David Fedida and mutagenesis was performed as described before [11]. All constructs were verified by Sanger sequencing.
Xenopus laevis oocytes were obtained by Ecocyte (Dortmund, Germany). Linearized cDNA was transcribed using the mMessage mMachine kit (Ambion) and the constructs were expressed in oocytes by injection of 5 to 50 ng human CLC-2 cRNA according to the expression level of the channel. Oocytes were kept in a Barth solution containing (in mM) 90 NaCl, 10 HEPES, 2 KCl, 1 MgCl2, 1 CaCl2, pH 7.5, for 3 days at 18°C. Two electrode voltage-clamp was performed with a Turbotec10 amplifier (npi, Tamm, npi electronic, Germany) and freeware acquisition program GePulse (freely available at http://users.ge.ibf.cnr.it/pusch/programs-mik.htm). The recordings were performed at room temperature (20°C). To analyse the currents of wild type CLC-2 and and mutant CLC-2, oocytes were first pulsed to 60 mV to estimate the instantaneous activated current under resting conditions, and then they were activated with a long pulse to –140 mV. The following tail pulse to 60 mV induced a very reproducible current deactivation, which can be best described by standard double exponential kinetics. The Imax of the tail pulse indirectly reflects the functional expression level, and the ratio Iss/Imax reflects the preference of the channel for the open configuration [37].
Human embryonic kidney (HEK) 293 cells were transfected with 1 µg human Kv1.5-eGFP cDNA (note: Kv1.5 is encoded by gene KCNA5) and recorded in extracellular solution containing (in mM): 140 NaCl, 2 MgSO4, 2 CaCl2 and 10 HEPES/NaOH pH 7.3 using the standard patch clamp technique. Intracellular solution was (in mM) 130 KCl, 2 MgSO4, 2 EGTA and 10 HEPES/NaOH pH 7.3. Voltage protocol and data analyses are as follows: Wild type and mutant hKv1.5 were elicited by 60 mV square pulses at a Vhold of -80 mV and current amplitudes were analyzed.
Neanderthal variant calling. The Neanderthal sequence contigs from six Neanderthal individuals (‘Feld1’, ‘Mez1’, ‘Sid1253’, ‘Vi33.16’, ‘Vi33.25’, ‘Vi33.26’) mapped against human hg18 reference genome was downloaded from Neandertal Genome Analysis Consortium Tracks at UCSC (ftp://hgdownload.cse.ucsc.edu/gbdb/hg18/neandertal/seqAlis/all-hg18.bam). SNP and short indel calling was performed using the UnifiedGenotyper algorithm implemented in GATK software suite [12-14]. ANNOVAR [15] was used to annotate the called variants with RefSeq gene names and to predict their impact and effect.
Mutation rate per gene has been calculated as variant count per length of human gene coding sequence (total SNVs) as well as functional variant count per length of human gene coding sequence (non-syn SNVs). Any non-synonymous coding variants or insertions/deletions have been considered for further analyses. Resulting mutation rates of ion channel genes have been log10-transformed to fit a normal distribution as tested by Shapiro-Wilk normality test implemented in R (p-value = 0.363). Mutation rates differing more than 2 standard deviations from mean of all analyzed ion channel genes have been considered as enriched mutation rates.
Results
By analyzing the published Neanderthal genomes and comparing them with 33 human disease-linked ion channel genes, we identified a high rate of genetic variation of uncertain significance (GVUC) in Neanderthal Cl- channel encoding genes CLCN2 and, to a lower degree, in CLCN1 (Fig. 1A, B). In AMHs, CLCN1 mutations associate with myotonia congenita, a rare disease characterized by muscle stiffness upon sudden forced movements [16, 17]. Regarding CLCN2, patients and knock-out mice with mutations in this gene show a more complex phenotype: CLC-2 dysfunction has been associated with retinal degeneration, leukodystrophy, and severe testicular degeneration [18-20]. Conversely, CLC-2 gain-of-function mutations have been shown to result in hyperaldosteronism type II; the increased aldosterone levels cause early-onset high blood pressure, predisposing affected individuals to potassium loss and potentially lethal cardiac arrhythmias [21, 22].
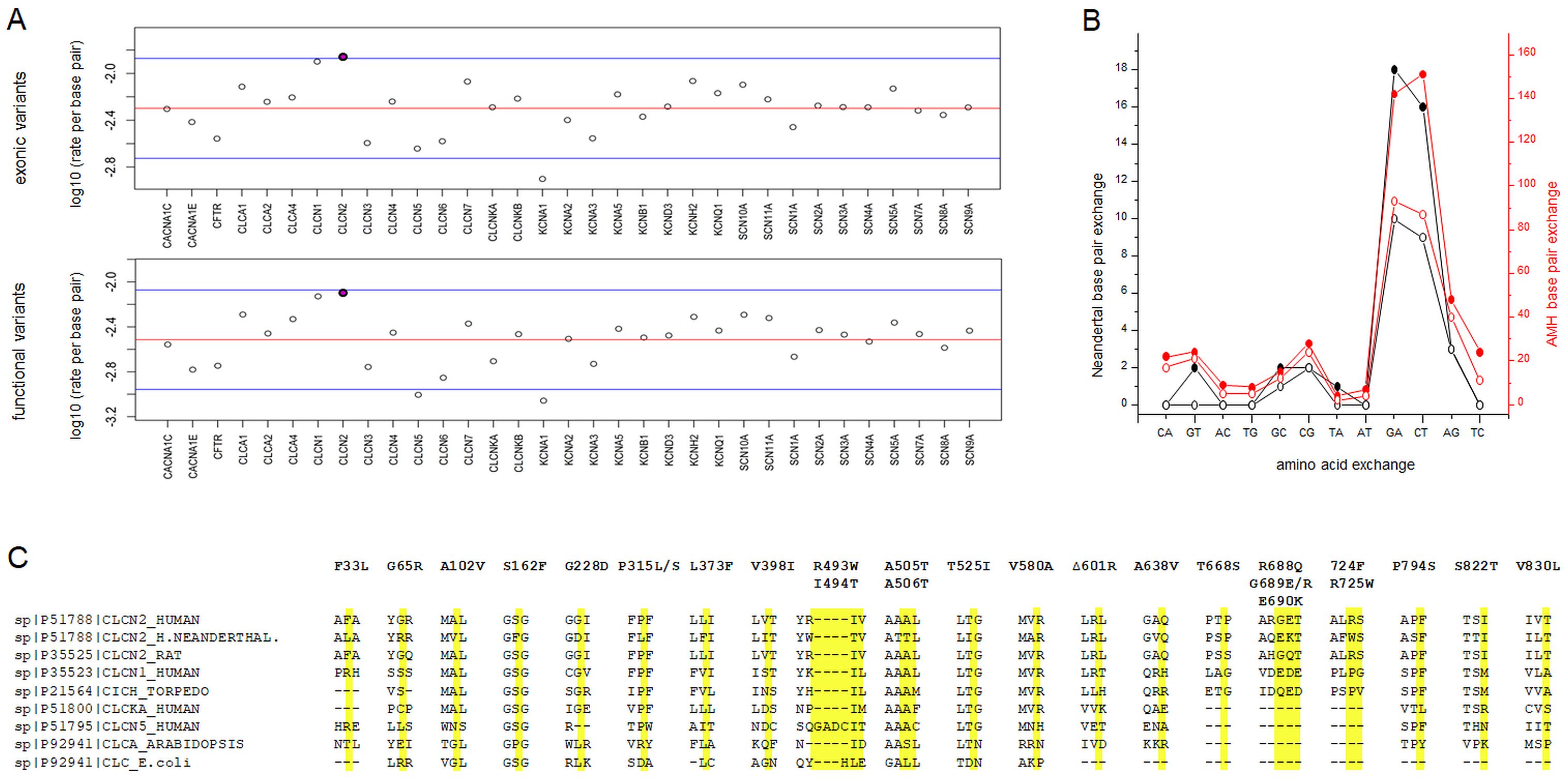
Typically, these GVUCs are filtered out in the process of genomic filtering leaving the genetic CLC-2 variations highly hypothetical. To exclude DNA degeneration as basis of Neanderthals genetic CLCN2 variation (Fig. 1A) we evaluated the pattern of base exchanges of Neanderthal gene variants. Neanderthal CLCN2 variants show a similar base pair exchange pattern compared to AMH, suggesting that we identified real variants rather than artifacts (Fig. 1B). Further, the CLCN2 sequence does not differ substantially in chemico-structural parameters (GC and AT content, intron/exon structure and length), meaning that the Neanderthal gene variants may have been present in living Neanderthals and potentially have impacted their lives (see below). The Neanderthal GVUCs are positioned in highly conserved regions of CLCN2, as shown by sequence alignments (Fig. 1C). CLCNx genes encode CLC-x proteins, a eukaryotic family of four Cl- anion channels (CLC-1, -2, -Ka, and -Kb) and five anion-H+ exchangers (CLC-3 to -7) containing 17 transmembrane α-helices [23]. CLCs are homo-dimers with separate active anion transport pathways in each subunit [24-27]. The structurally very similar prokaryotic and eukaryotic CLC anion-H+ exchangers have been crystallized [24, 28, 29], and we used these as templates to generate a structural model of CLC-2. With this model, we localized the position of several of the variants to highly sensitive regions of the CLC-2 channel protein: the transmembrane domain, which includes the selectivity filter and intracellular CBS domain (Fig. 2B). These findings suggest that the gene variants could result in impaired Cl- ion channel function.
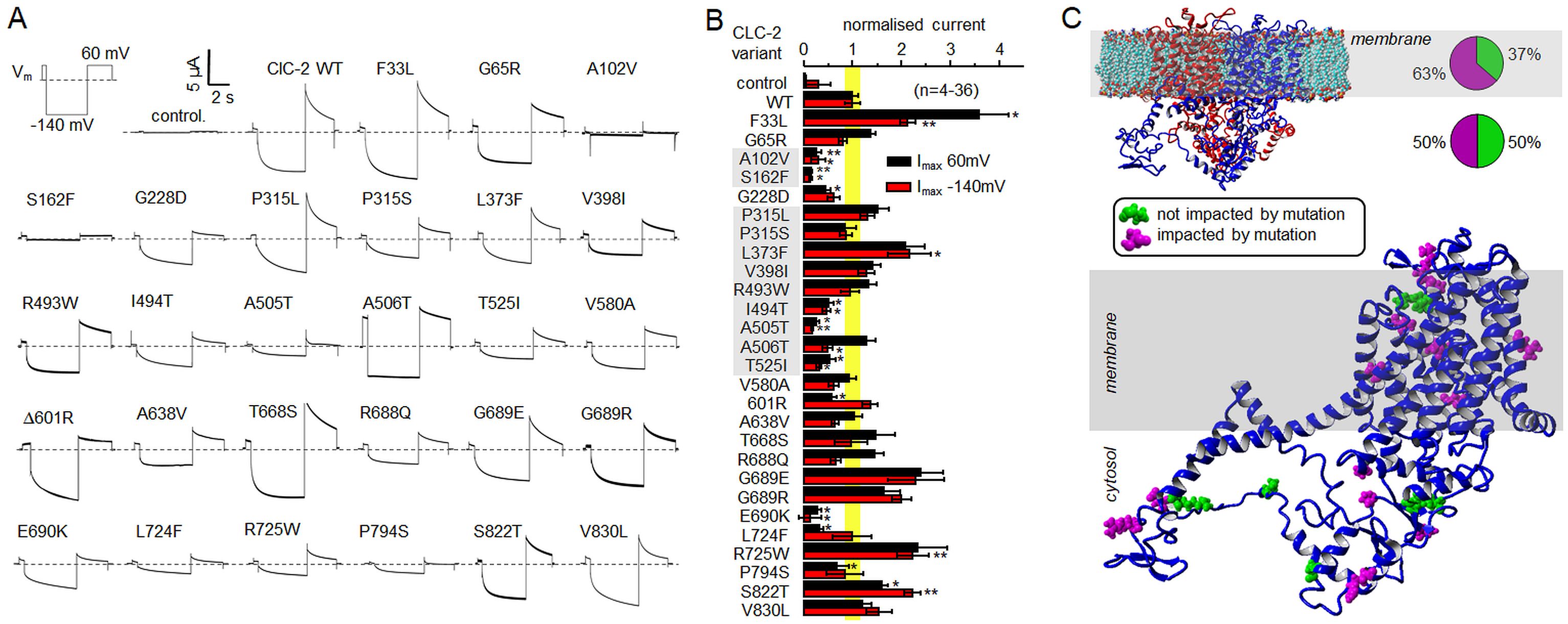
In order to address the functional effects of the respective gene variants directly, we generated individual Neanderthal CLCN2 gene variants and analyzed them functionally in Xenopus laevis oocytes (Fig. 2A, B). We found diverse functional effects as a result of the various gene variants.
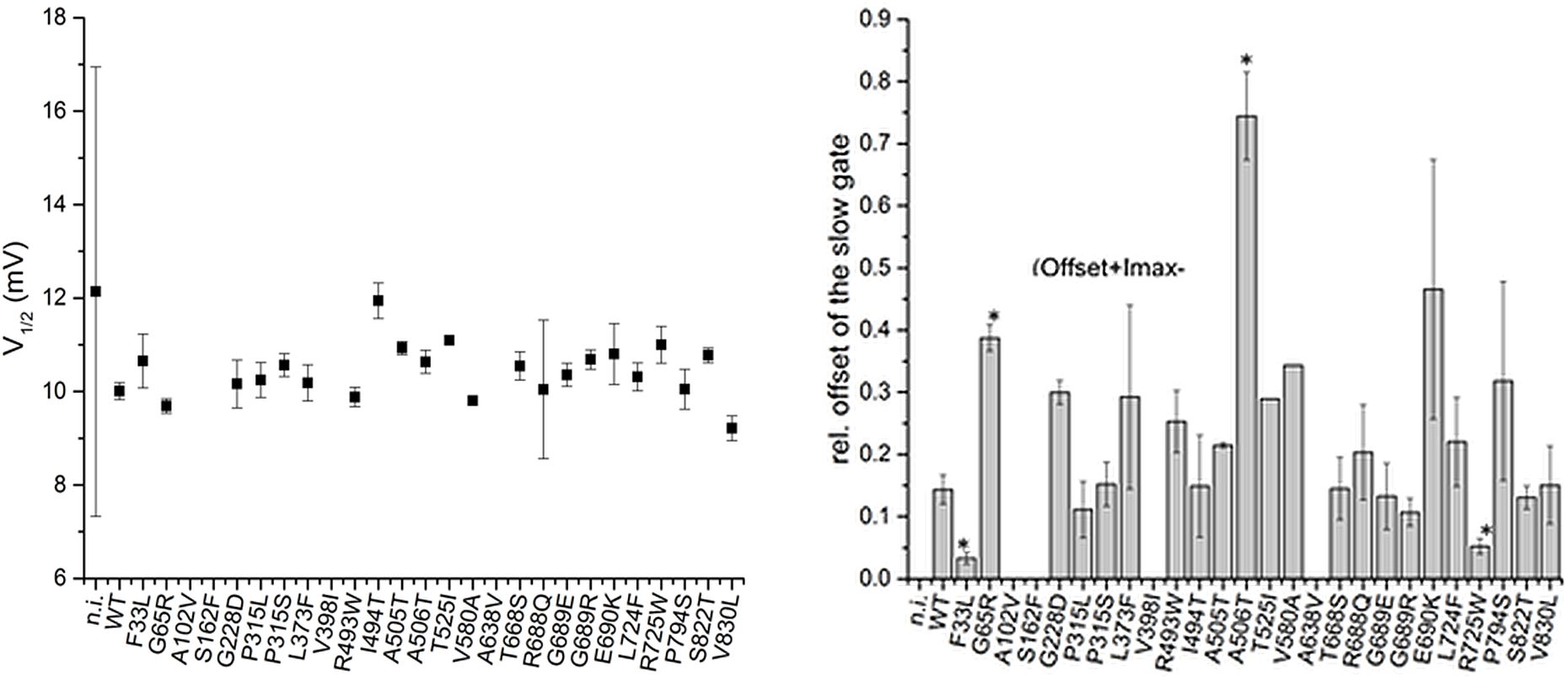
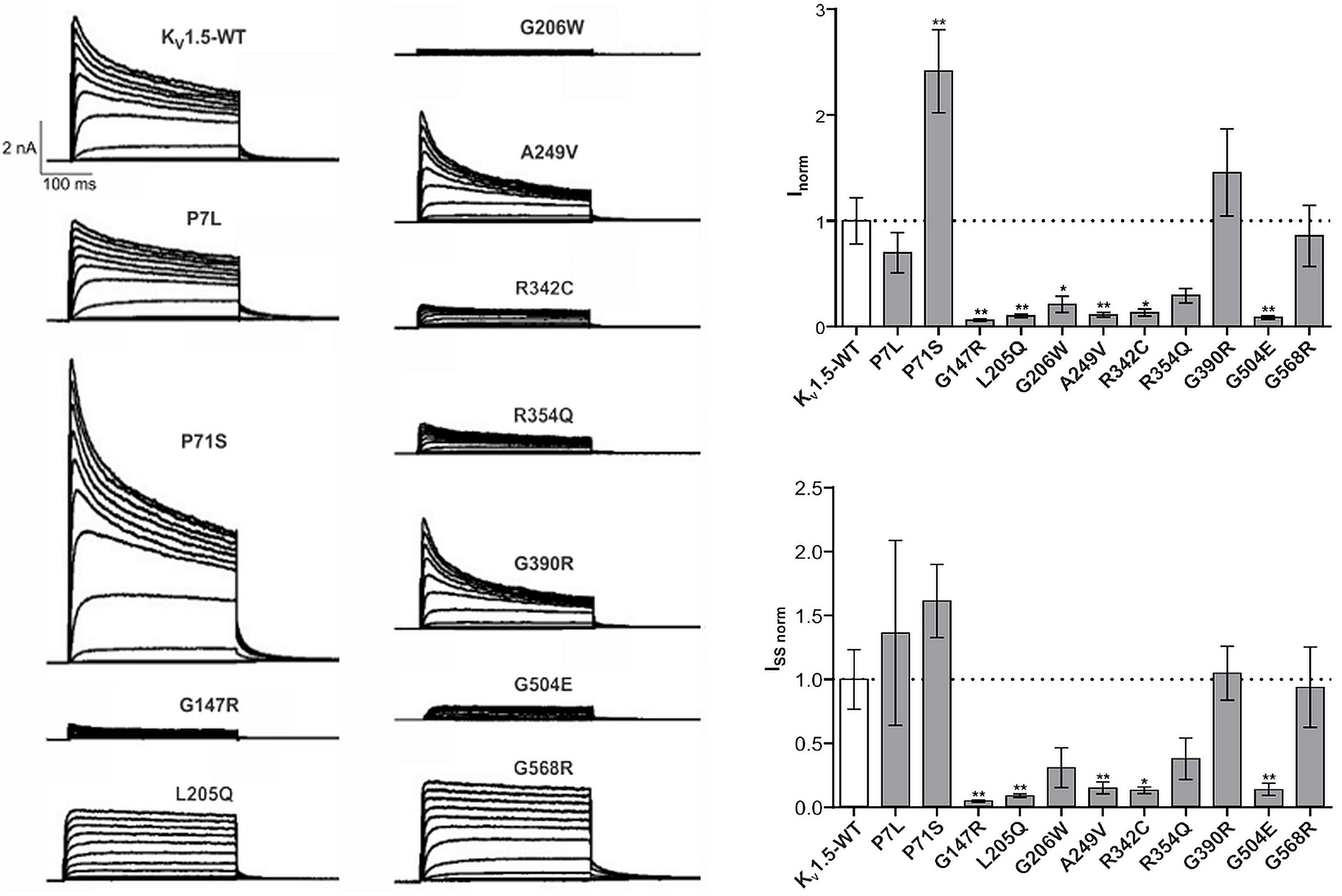
CLC-2 activates upon hyperpolarization and may contribute a Cl- efflux pathway to regulate the basolateral Cl- efflux in epithelial cells [30-32]. We found that the steady state CLC-2 currents were reduced by eleven variants and were increased by four variants. Further, CLC-2 channel gating is characterized by two processes that open the channel at negative voltages, reflecting opening of individual protopores (fast gate), and a slower mechanism that operates on both pores of the double-barreled channel [33-36]. Conversely, at positive voltages, the channel reaches its minimum open probability, and this probability can be affected by mutation [36] or by association with the glial-specific cell adhesion molecule GlialCAM [37, 38]. We found that four variant channels showed changes in gating kinetics and altered the minimum open probability (Fig. 3). Thus, overall 59.3 % of the potential Neanderthal variants resulted in functional aberrations (41 % with loss-of-function and 15 % with gain-of-function, and 15% showed gating abnormalities, Fig. 3). To determine if the percentage of CLCN2 GVUCs that we identified as having functional consequences (59.3%) coincides with this percentage for other Neanderthal ion channel genes, we examined potential Neanderthal variants of the alternative ion channel Kv1.5 (Fig. 4): 63.6 % (7 out of 11) of Kv1.5 Neanderthal variants showed significant functional changes, suggesting that the number of Neanderthal gene variants correlates with the number of functionally relevant gene variants (Fig. 4).
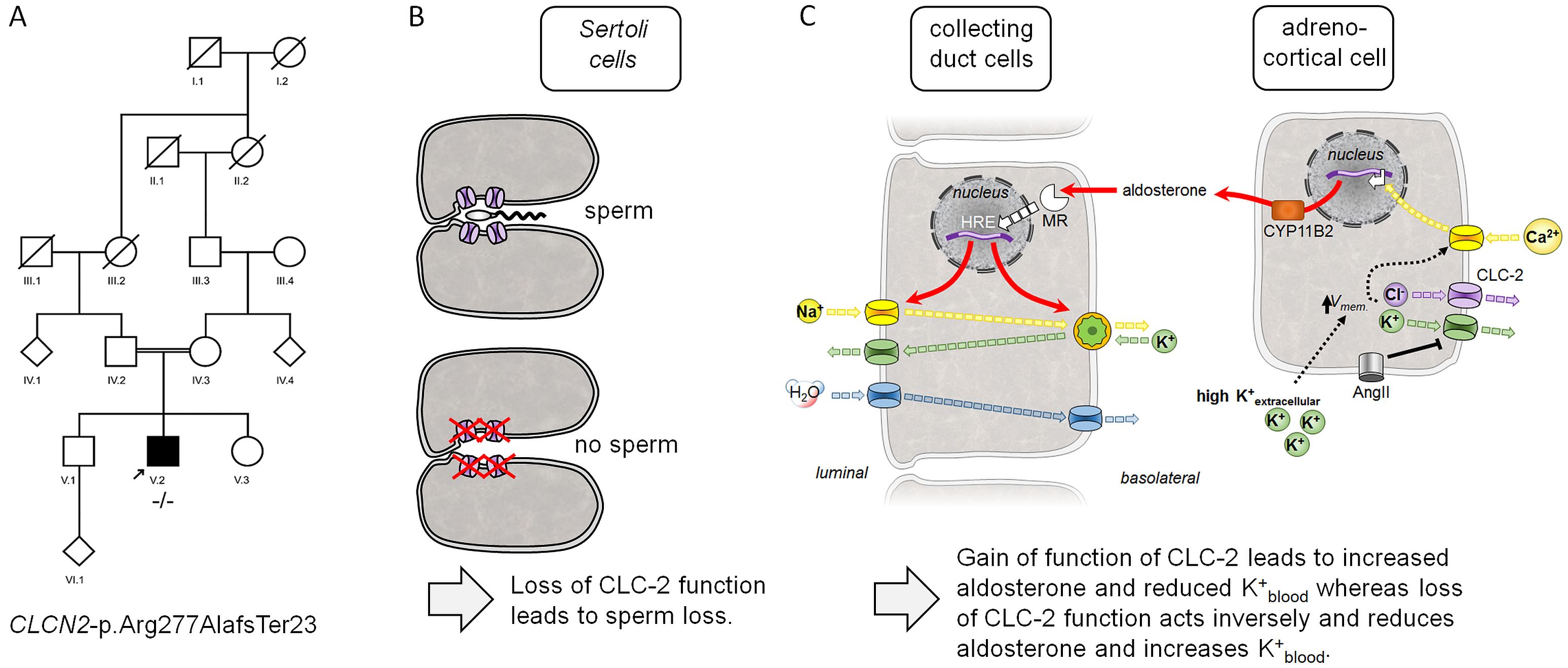
Human loss-of-function mutations in CLCN2 have been associated with leukodystrophy [18] and azoospermia [19], suggesting that CLC-2 dysfunction can cause male infertility [39]. Consistently, CLC-2 knock-out mice also exhibited testicular degeneration, whereby their primary spermatocytes and later the spermatogonia degenerated, and the tubules contained abnormal Sertoli cells [39]. In order to verify the suggested critical role for CLC-2 function in normal spermatogenesis, we screened whole-exome sequencing data of 887 infertile men with severe spermatogenic failure (azoo-, crypto- or severe oligozoospermia) from the Male Reproductive Genomics (MERGE) cohort for rare variants of the CLCN2 gene. A 31-year-old otherwise healthy male (M1374) carried the homozygous CLCN2 mutation c.828dup (NM_004366.5) leading to frameshift p.Arg277Alafs and termination of the protein at residue 23 (Fig. 5A). No other genetic variant potentially explaining his azoospermia was identified in 194 genes previously associated with spermatogenic failure. A testicular biopsy was performed, and testicular sperm extraction (TESE) yielded no sperm. Histological analysis revealed that most tubuli showed germ cell loss (Sertoli cell-only phenotype), and only a few tubules showed residual spermatogenesis but only up to spermatocytes, suggesting arrest of spermatogenesis mimicking the murine phenotype [39]. MRI of the patient was not available.
Discussion
Previous reports and the findings presented here underscore that CLC-2 has a key role in normal sperm production. Based on the critical role of CLC-2 in spermatogenesis, the Neanderthal CLCN2 gene variants, which reduce CLC-2 function in heterologous expression, would likely also lead to reduced production of competent sperm. As a result, reduced sperm number may be expected to result in reduced reproductive success. In line with this hypothesis, Neanderthal populations were known to have small group sizes and low genetic diversity. Supposedly, these characteristics were facilitated by Neanderthals patrilocal mating behavior, which would have focused reproductive success on the Neanderthal males [9].
Two recent Nature Genetics studies have shown that gain-of-function in CLC-2 leads to hyperaldosteronism type II [21, 22]. Inversely, mild CLC-2 loss-of-function may have resulted in lower aldosterone levels, lower blood pressure, and potassium-saving. Indeed, the CLC-2 loss of function patient analyzed here (Fig. 5A) featured high serum potassium levels of 5.1 mmol/l (3.5-5.0), underscoring a proposed potassium-saving mechanism (Fig. 5C): CLC-2 function determines aldosterone levels, which in turn regulate sodium vs. potassium exchange in the renal collecting duct (Fig. 5C). Typical diets in the Stone Age included a 4-fold higher potassium uptake than in current diet due to consuming various plants (fruits, leafy greens, vegetable fruits, and roots) and high NaCl uptake [40]. The combination of a high-potassium diet and a salt-rich diet would be expected to lead to increased aldosterone levels and, potentially, to increased blood pressure. Thus, a reduced CLC-2 function could counteract these effects by decreasing aldosterone secretion; the Neanderthal CLC-2-gene loss-of-function variants might reflect an adaptation to salt-rich diet. On the other hand, for Neanderthals living in regions with a low salt supply, increased CLC-2 function due to gain-of-function gene variants would allow for highly efficient reabsorption of NaCl in the kidneys. Thus, depending on the environment, a loss or gain of function in CLC-2 could be expected to be beneficial. Therefore, it is plausible that Neanderthal populations with variable CLC-2 functions may have been better prepared for local NaCl abundances and different local diets.
Conclusion
In summary, Neanderthal genomes exhibit an unusually high density of CLCN2 gene variants, which may potentially indicate technical artifacts. However, physicochemical features of these variants are similar to AMH and functional analyses of the Neanderthals gene variants show that they impair Cl- channel function in most cases. Potentially, this could represent a potential adaptation to local salt supply and could have also been associated with reduced fertility in male Neanderthals. Current rare loss-of-function gene variants in CLCN2 contribute to azoospermia and male infertility in modern humans.
Author Contributions
Elena Jeworutzki, Ina Rothenberg, Frank Tüttelmann, Michael Pusch, Julian A. Schreiber, Sabine Kliesch and Guiscard Seebohm collected data and performed analyses; Guiscard Seebohm designed the study; Elena Jeworutzki, Frank Tüttelmann and Guiscard Seebohm prepared figures; Frank Tüttelmann, Michael Pusch, Bernhard Wünsch, Nathalie Strutz-Seebohm and Guiscard Seebohm wrote the manuscript. We thank Frank Rühle, Andrei Barysenka and Monika Stoll with help calling Neanderthal gene variants and Albrecht Röpke who helped with the genetic workup of the infertile patient.
Funding
Parts of this study were supported by the German Research Foundation sponsored Clinical Research Unit ‘Male Germ Cells: from Genes to Function’ (DFG CRU326, grants to FT) and the German Research Foundation Graduate school Chembion (to GS and BW). Research of MP is funded by grants from the Fondazione AIRC per la Ricerca sul Cancro (grant # IG 21558) and the Italian Research Ministry (PRIN 20174TB8KW).
The authors declare that no conflict of interests exists.
| 1 Sankararaman S, Patterson N, Li H, Paabo S, Reich D: The Date of Interbreeding between Neandertals and Modern Humans. PLoS Genet 2012;8:e1002947. https://doi.org/10.1371/journal.pgen.1002947 |
||||
| 2 Benazzi S, Slon V, Talamo S, Negrino F, Peresani M, Bailey SE, Sawyer S, Panetta D, Vicino G, Starnini E, Mannino MA, Salvadori PA, Meyer M, Paabo S, Hublin JJ: The makers of the Protoaurignacian and implications for Neandertal extinction. Science 2015;348:793-796. https://doi.org/10.1126/science.aaa2773 |
||||
| 3 Dalen L, Orlando L, Shapiro B, Brandstrom-Durling M, Quam R, Gilbert MTP, Fernandez-Lomana JCD, Willerslev E, Arsuaga JL, Gotherstrom A: Partial Genetic Turnover in Neandertals: Continuity in the East and Population Replacement in the West. Mol Biol Evol 2012;29:1893-1897. https://doi.org/10.1093/molbev/mss074 |
||||
| 4 Maricic T, Gunther V, Georgiev O, Gehre S, Curlin M, Schreiweis C, Naumann R, Burbano HA, Meyer M, Lalueza-Fox C, de la Rasilla M, Rosas A, Gajovic S, Kelso J, Enard W, Schaffner W, Paabo S: A Recent Evolutionary Change Affects a Regulatory Element in the Human FOXP2 Gene. Mol Biol Evol 2013;30:844-852. https://doi.org/10.1093/molbev/mss271 |
||||
| 5 Houldcroft CJ, Underdown SJ: Neanderthal genomics suggests a pleistocene time frame for the first epidemiologic transition. Am J Phys Anthropol 2016;160:379-388. https://doi.org/10.1002/ajpa.22985 |
||||
| 6 Lalueza-Fox C, Gilbert MTP, Martinez-Fuentes AJ, Calafell F, Bertranpetit J: Mitochondrial DNA from pre-Columbian Ciboneys from Cuba and the prehistoric colonization of the Caribbean. Am J Phys Anthropol 2003;121:97-108. https://doi.org/10.1002/ajpa.10236 |
||||
| 7 Khrameeva EE, Bozek K, He L, Yan Z, Jiang X, Wei YN, Tang K, Gelfand MS, Prufer K, Kelso J, Paabo S, Giavalisco P, Lachmann M, Khaitovich P: Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans. Nat Commun 2014;5:3584. https://doi.org/10.1038/ncomms4584 |
||||
| 8 Williams AL, Jacobs SBR, Moreno-Macias H, Huerta-Chagoya A, Churchhouse C, Marquez-Luna C, Garcia-Ortiz H, Gomez-Vazquez MJ, Burtt NP, Aguilar-Salinas CA, Gonzalez-Villalpando C, Florez JC, Orozco L, Haiman CA, Tusie-Luna T, Altshuler D, Williams AL, Marquez-Luna C, Huerta-Chagoya A, Ripke S, et al.: Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 2014;506:97-101. https://doi.org/10.1038/nature12828 |
||||
| 9 Lalueza-Fox C, Rosas A, Estalrrich A, Gigli E, Campos PF, Garcia-Tabernero A, Garcia-Vargas S, Sanchez-Quinto F, Ramirez O, Civit S, Bastir M, Huguet R, Santamaria D, Gilbert MTP, Willerslev E, de la Rasilla M: Genetic evidence for patrilocal mating behavior among Neandertal groups. Proc Natl Acad Sci U S A 2011;108:250-253. https://doi.org/10.1073/pnas.1011553108 |
||||
| 10 Mellars P, French JC: Tenfold Population Increase in Western Europe at the Neandertal-to-Modern Human Transition. Science 2011;333:623-627. https://doi.org/10.1126/science.1206930 |
||||
| 11 Strutz-Seebohm N, Henrion U, Schmitt N, Schulze-Bahr E, Seebohm G: A Common Structural Component for beta-Subunit Mediated Modulation of Slow Inactivation in Different K-V Channels. Cell Physiol Biochem 2013;31:968-980. https://doi.org/10.1159/000350115 |
||||
| 12 DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491-498. https://doi.org/10.1038/ng.806 |
||||
| 13 McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA: The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297-1303. https://doi.org/10.1101/gr.107524.110 |
||||
| 14 Van der Auwera GA, Carneiro MO HC, Poplin R DAG, Levy-Moonshine A, Jordan T SK, Roazen D TJ, Banks E GK, Altshuler D GS, MA D: From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1-11.10.33. https://doi.org/10.1002/0471250953.bi1110s43 |
||||
| 15 Wang K, Li MY, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. https://doi.org/10.1093/nar/gkq603 |
||||
| 16 Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmannhorn F, Grzeschik KH, Jentsch TJ: The Skeletal-Muscle Chloride Channel in Dominant and Recessive Human Myotonia. Science 1992;257:797-800. https://doi.org/10.1126/science.1379744 |
||||
| 17 Lossin C, George AL: Myotonia Congenita. Adv Genet 2008;63:25-55. https://doi.org/10.1016/S0065-2660(08)01002-X |
||||
| 18 Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, van Berkel C, Polder E, Tollard E, Darios F, Brice A, de Die-Smulders CE, Vles JS, Vanderver A, Uziel G, Yalcinkaya C, Frints SG, Kalscheuer VM, Klooster J, Kamermans M, et al.: Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013;12:659-668. https://doi.org/10.1016/S1474-4422(13)70053-X |
||||
| 19 Di Bella D, Pareyson D, Savoiardo M, Farina L, Ciano C, Caldarazzo S, Sagnelli A, Bonato S, Nava S, Bresolin N, Tedeschi G, Taroni F, Salsano E: Clinical/Scientific Notes. Neurology 2014;83:1217-1218. https://doi.org/10.1212/WNL.0000000000000812 |
||||
| 20 Hoegg-Beiler MB, Sirisi S, Orozco IJ, Ferrer I, Hohensee S, Auberson M, Godde K, Vilches C, de Heredia ML, Nunes V, Estevez R, Jentsch TJ: Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun 2014;5:3475. https://doi.org/10.1038/ncomms4475 |
||||
| 21 Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, Boulkroun S, Jeunemaitre X, Amar L, Lefebvre H, Schwarzmayr T, Strom TM, Jentsch TJ, Zennaro MC: A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet 2018;50:355-361. https://doi.org/10.1038/s41588-018-0053-8 |
||||
| 22 Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, Jin SC, Loring E, Untiet V, Yoo T, Choi J, Xu SX, Wu AH, Kirchner M, Mertins P, Rump LC, Onder AM, Gamble C, McKenney D, et al.: CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 2018;50:349-354. https://doi.org/10.1038/s41588-018-0048-5 |
||||
| 23 Ponting CP: CBS domains in ClC chloride channels implicated in myotonia and nephrolithiasis (kidney stones). J Mol Med (Berl) 1997;75:160-163. | ||||
| 24 Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R: X-ray structure of a CIC chloride channel at 3.0 angstrom reveals the molecular basis of anion selectivity. Nature 2002;415:287-294. https://doi.org/10.1038/415287a |
||||
| 25 Jentsch TJ, Pusch M: Clc Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol Rev 2018;98:1493-1590. https://doi.org/10.1152/physrev.00047.2017 |
||||
| 26 Miller C: Open-State Substructure of Single Chloride Channels from Torpedo Electroplax. Philos Trans R Soc Lond B Biol Sci 1982;299:401-411. https://doi.org/10.1098/rstb.1982.0140 |
||||
| 27 Miller C, White MM: Dimeric Structure of Single Chloride Channels from Torpedo Electroplax. Proc Natl Acad Sci U S A 1984;81:2772-2775. https://doi.org/10.1073/pnas.81.9.2772 |
||||
| 28 Dutzler R, Campbell EB, MacKinnon R: Gating the selectivity filter in ClC chloride channels. Science 2003;300:108-112. https://doi.org/10.1126/science.1082708 |
||||
| 29 Feng LA, Campbell EB, Hsiung YC, MacKinnon R: Structure of a Eukaryotic CLC Transporter Defines an Intermediate State in the Transport Cycle. Science 2010;330:635-641. https://doi.org/10.1126/science.1195230 |
||||
| 30 Catalan M, Cornejo I, Figueroa CD, Niemeyer MI, Sepulveda FV, Cid LP: ClC-2 in guinea pig colon: mRNA, immunolabeling, and functional evidence for surface epithelium localization. Am J Physiol Gastrointest Liver Physiol 2002;283:G1004-G1013. https://doi.org/10.1152/ajpgi.00158.2002 |
||||
| 31 Catalan MA, Flores CA, Gonzalez-Begne M, Zhang Y, Sepulveda FV, Melvin JE: Severe Defects in Absorptive Ion Transport in Distal Colons of Mice That Lack ClC-2 Channels. Gastroenterology 2012;142:346-354. https://doi.org/10.1053/j.gastro.2011.10.037 |
||||
| 32 Zdebik AA, Cuffe JE, Bertog M, Korbmacher C, Jentsch TJ: Additional disruption of the ClC-2Cl(-) channel does not exacerbate the cystic fibrosis phenotype of cystic fibrosis transmembrane conductance regulator mouse models. J Biol Chem 2004;279:22276-22283. https://doi.org/10.1074/jbc.M309899200 |
||||
| 33 de Santiago JA, Nehrke K, Arreola J: Quantitative analysis of the voltage-dependent gating of mouse parotid ClC-2 chloride channel. J Gen Physiol 2005;126:591-603. https://doi.org/10.1085/jgp.200509310 |
||||
| 34 Niemeyer MI, Cid LP, Yusef YR, Briones R, Sepulveda FV: Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J Physiol 2009;587:1387-1400. https://doi.org/10.1113/jphysiol.2008.167353 |
||||
| 35 Thiemann A, Grunder S, Pusch M, Jentsch TJ: A Chloride Channel Widely Expressed in Epithelial and Nonepithelial Cells. Nature 1992;356:57-60. https://doi.org/10.1038/356057a0 |
||||
| 36 Zuniga L, Niemeyer MI, Varela D, Catalan M, Cid LP, Sepulveda FV: The voltage-dependent CIC-2 chloride channel has a dual gating mechanism. J Physiol 2004;555:671-682. https://doi.org/10.1113/jphysiol.2003.060046 |
||||
| 37 Jeworutzki E, Lagostena L, Elorza-Vidal X, Lopez-Hernandez T, Estevez R, Pusch M: GlialCAM, a CLC-2 Cl- Channel Subunit, Activates the Slow Gate of CLC Chloride Channels. Biophys J 2014;107:1105-1116. https://doi.org/10.1016/j.bpj.2014.07.040 |
||||
| 38 Jeworutzki E, Lopez-Hernandez T, Capdevila-Nortes X, Sirisi S, Bengtsson L, Montolio M, Zifarelli G, Arnedo T, Muller CS, Schulte U, Nunes V, Martinez A, Jentsch TJ, Gasull X, Pusch M, Estevez R: GlialCAM, a Protein Defective in a Leukodystrophy, Serves as a CIC-2 Cl- Channel Auxiliary Subunit. Neuron 2012;73:951-961. https://doi.org/10.1016/j.neuron.2011.12.039 |
||||
| 39 Bosl MR, Stein V, Hubner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, Davidoff MS, Holstein AF, Jentsch TJ: Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2Cl(-) channel disruption. EMBO J 2001;20:1289-1299. https://doi.org/10.1093/emboj/20.6.1289 |
||||
| 40 Sebastian A, Frassetto LA, Sellmeyer DE, Morris RC: The evolution-informed optimal dietary potassium intake of human beings greatly exceeds current and recommended intakes. Semin Nephrol 2006;26:447-453. https://doi.org/10.1016/j.semnephrol.2006.10.003 |
||||