×
![]()
Corresponding Author: Jiro Fujimoto
Hyogo College of Medicine, Department of Gastroenterological Surgery,
Nishinomiya 663-8501, Hyogo (Japan)
Tel. +81-798-45-6582, E-Mail sfujimo@hyo-med.ac.jp
Antithrombin Together with NETs Inhibitor Protected Against Postoperative Adhesion Formation in Mice
Makoto Sudo Jinyang Xu Keiko Mitani Mayo Jimbo Hiroko Tsutsui Etsuro Hatano Jiro Fujimoto
Department of Gastroenterological Surgery, Hyogo College of Medicine, Nishinomiya, Hyogo, Japan
Introduction
Laparotomy frequently results in peritoneal adhesion formation, which can induce secondary illnesses such as chronic abdominal pain, small bowel obstruction, infertility in women, and difficulties in subsequent surgical interventions [1-4]. To reduce the burden of postoperative adhesions, laparoscopic surgery, instead of laparotomy, has spread rapidly and can reduce the incidence of adhesion-related readmissions. However, postoperative adhesion remains a major clinical issue. Some preventive medications such as barrier sheets preventing contact between the surgically injured sites and the other healthy peritoneal organs, have been established [5, 6]. However, adhesion does not only occur in the macro-injured region, but may also develop at any sites with micro injuries induced by the surgery itself. The limited effects of the physical barrier sheets prompted us to develop anti-adhesive medications for systemic administration.
Recently, we showed that blockade of TNF or IL-6 signaling could prevent adhesion formation induced by cauterization of the cecum [7, 8]. In surgically injured serosa, robust neutrophils promptly accumulate and produce neutrophil extracellular traps (NETs), a web-like structure consisting of neutrophil-derived molecules, while blockade of these cytokines prevents neutrophilia and NET production [7, 8]. Furthermore, in vivo depletion of neutrophils rescues mice from adhesion formation [7]. Taken together these observations suggest the importance of neutrophils in the development of adhesions. NETs were originally reported as an innate immune response designed to reduce microbial infection [9]. In their resting state, neutrophils safely store various types of harmful enzymes in their cytoplasmic granules. However, upon NET release, these granular enzymes together with other neutrophil-derived molecules, such as histones and DNA, are liberated into the extracellular space, which eventually kill the microbes [9-11]. The same mechanisms for NET-mediated microbial killing, however, can insult host tissues whenever targeting the healthy host cells. In fact, many reports have shown that NETs are also involved in the production and development of diseases [10-14]. This allowed us to assume that NET release accounts for some of the postoperative adhesion formation.
We previously reported fibrin deposition and expression of plasminogen activator inhibitor 1 (PAI-1), which is a potent anti-fibrinolytic and procoagulant molecule [15], in the injured serosa of mice receiving intraperitoneal surgeries such as cecum cauterization and partial hepatectomy [7, 8]. In fact, dense fibrin deposition, an end product of coagulation, also develops in postoperative humans [7]. Blockade or genetic depletion of PAI-1 prevents adhesion formation [16, 17], suggesting the importance of this procoagulant in adhesion production.
Thrombin is a key molecule in the hemostatic process [18]. Thrombin activates platelets and converts fibrinogen into fibrin, leading to the formation of stable fibrin clots. Antithrombin (AT) is a natural inhibitor of thrombin [19-21]. Intriguingly, procoagulants induce NET release [22-24] and NET release can promote the procoagulant state [10, 11, 25, 26]. Thus, NET formation and procoagulant expression seem to work in a positive feedback loop, potentially aggravating adhesion production. This may suggest that inhibition of either process might result in partial protection against adhesion formation. Moreover, AT administered could not fully exert its anticoagulant activity under conditions of NET release, because elastase liberated by the NET release, might inactivate and degrade AT [27, 28]. These prompted us to propose the hypothesis that adhesions may only be eliminated completely when both pathways are ablated. Here, we investigate whether AT or NET inhibitors can rescue mice who receive cecum cauterization from adhesion formation, and if this is the case, establish to what extent the combination of treatments against both pathways improved therapeutic response.
Materials and Methods
Reagents
Freeze-dried, concentrated human antithrombin III was kindly provided by the Japanese Blood Products Organization (JB). Thrombin from bovine plasma (T-4648) was purchased from Sigma, GSK484 (hydrochloride) (#17488) from Cayman Chemical., anti-Ly6G mAb (551459) from BD Biosciences, and anti-histone H3 (citrulline R2 + R8 + R17) antibody-ChIP Grade (ab5103) from Abcam.
Mice
In the cecum cauterization model, there were no differences in adhesion production in terms of clinical score or histological features between male and female mice [7, 8, 16, 17]. Given this, female BALB/c mice (8–10 weeks of age) were purchased from Japan SLC, Inc. All animal experiments were performed under specific pathogen-free conditions in accordance with the guidelines of the Institutional Animal Care Committee of Hyogo College of Medicine (protocol number: 17-019).
Murine cecum cauterization
We previously described a mouse cecum-cauterization model [7, 8, 16]. Briefly, we anesthetized mice using isoflurane which was followed by a midline abdominal incision and cauterization of the cecum isolated using the coagulation mode on a set of bipolar forceps (MERA MS-1500; output dial 4; 5 W, 500kHz, 100 Ω Senko Medical Instrument Manufacturing CO, Japan) with a pinch width of 0.3 mm for 2 s. The incision was closed in two layers using silk sutures and the injured ceca and its surrounding tissues were sampled at the relevant time points. On day 7 post-surgery, adhesion formation was scored from 0 to 5 according to the following scoring system: 0, no adhesion; 1, one thin filmy adhesion; 2, more than one thin adhesion; 3, thick adhesion with focal point; 4, thick adhesion with plantar attachment or more than one thick adhesion with focal point; and 5, very thick vascularized adhesion or more than one plantar adhesion. In some experiments, mice were intraperitoneally treated with AT and/or GSK484 prior to and/or post-operation.
Tissue staining
Hematoxylin and eosin (HE) and Sirius red staining were performed as previously described [8].
Immunohistochemistry
Immunohistochemical analysis was performed as previously described [8]. Briefly, we fixed tissue specimens in IHC Zinc Fixative (BD Pharmingen) and embedded them in paraffin wax before sectioning. The sections were preincubated with a serum-free protein block (Agilent Technologies) for 30 min at room temperature and the primary antibodies were added to each slide. Secondary antibody staining was then performed using Histofine Simple Stain Mouse MAX-PO (Nichirei Biosciences Inc.) according to the manufacturer’s instructions. The staining was then visualized under a Nikon Eclipse Ni-U microscope (Nikon). In some experiments, we measured the histochemical intensity using ImageJ software version 1.52a (SciJava software ecosystem’s open source software project) [7, 8].
Immunofluorescence staining with confocal microscopy
Immunofluorescence analysis was performed as previously described [8]. Briefly, tissue samples were fixed in IHC Zinc Fixative and embedded in paraffin wax. Fibrosis was detected by incubating the tissue slices with antibodies against a-SMA (ab124964, Abcam) and podoplanin (ab11936, Abcam) or 4’,6-diamidino-2-phenylindole (DAPI) to detect DNA. To detect neutrophil extracellular traps (NETs), the slices were incubated with antibodies against Ly6G (551459 BD Bioscience), anti-citrullinated histone H3 mAbs, and DAPI. Fluorescence was visualized under a confocal microscope (Zeiss LSM780).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as previously described [8]. Total RNA was prepared from tissue or cell samples using an RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA), and reverse transcripts were synthesized using PrimeScriptTM RT Master Mix (Perfect Real Time) (Takara Bio, Inc.). TaqMan Fast Advanced Master Mix, TaqMan gene expression assays for Il6 (Mm00446190_m1), Tnf (Mm00443258_m1), Pai1 (Mm00435858_m1), Cxcl2 (Mm00436450_m1), IL6 (Hs00174131_m1), TNF (Hs00174128_m1), CXCL2 (Hs00601975_m1), and 18S (Hs99999901_s1) were purchased from Applied Biosystems. qPCR was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). The results were analyzed using StepOne software v.2.0 and the fold-change in each gene was analyzed using the 2−ΔΔCt method. According to this method, each threshold cycle (Ct) value was first normalized to the internal reference gene (18S ribosomal RNA) in each sample and then compared to the control.
Culture of mesothelial cells
MeT-5A cells (3 × 105 cells) were seeded in 3.5 cm dishes in Medium 199 (Gibco) containing 10% FBS and incubated with thrombin with or without AT for 24 h at 37°C and 5% CO2.
Statistical analyses
Data are described as the mean ± SEM of either the results from each experimental group or triplicate samples. Statistical comparisons were performed using Tukey’s tests with free web software (www.gen-info.osaka-u.ac.jp/MEPHAS/tukey.html) [8] or Student’ s t-test. Statistical significance was set at P < 0.05.
Results
AT partially prevents the development of postoperative adhesions
Thrombin is a key enzyme in fibrin clot generation, facilitating the activation of the platelets and the conversion of fibrinogen to fibrin [18]. To investigate the importance of this procoagulant state in postoperative adhesions, we administered AT to mice day 1 and 1 h before and once a day for 5 consecutive days after cauterization. Mice treated with AT exhibited significantly reduced adhesion scores compared to those in the control group that were treated with PBS (Fig. 1A). The PBS-treated control mice developed thick, vascularized adhesions while the AT treated group developed only thick adhesions (Fig. 1A). We then went on to test whether prophylactic treatment with AT could prevent the development of adhesions in mice. To do this, we administered AT twice, once at 1 d and once at 1 h prior to cauterization (Pre). In contrast to the full-term administration (Full), preadministration of AT did not suppress adhesion formation (Supplementary Fig. 1 – for all supplementary material see www.cellphysiolbiochem.com). Therefore, we administered AT using the full-term protocol. Immunohistochemistry against thrombin and fibrin/fibrinogen revealed that AT-treated mice produced a lower number of cells expressing fibrin in the injured serosa than the control (Fig. 1B). Thus, AT-mediated protection against adhesions seems to be associated with the impairment of the procoagulant state.
Recent studies have revealed an additional role for proinflammatory features in the development of adhesions. These studies suggest that proinflammatory factors activate endothelial and immune cells increasing their production of leukocyte-recruiting molecules and proinflammatory cytokines, such as IL-6 [29-34]. In fact, we found that human mesothelial cells, which are a major cellular source of the myofibroblasts that make up the adhesion band [7, 35], express IL6 and PAI1 (Fig. 1C). Interestingly, AT treatment directly inhibited the thrombin-mediated induction of these transcripts (Fig. 1C). This prompted us to investigate whether AT treatment prevents inflammatory responses in the injured serosa of cecal cauterized mice. Unexpectedly, AT did not protect against regional inflammatory responses, such as the induction of Il6, Tnf, Pai1, or Cxcl2 (Fig. 1D) [8]. These results suggest that AT treatment inhibit the development of postoperative adhesions without modulating the local inflammatory responses.
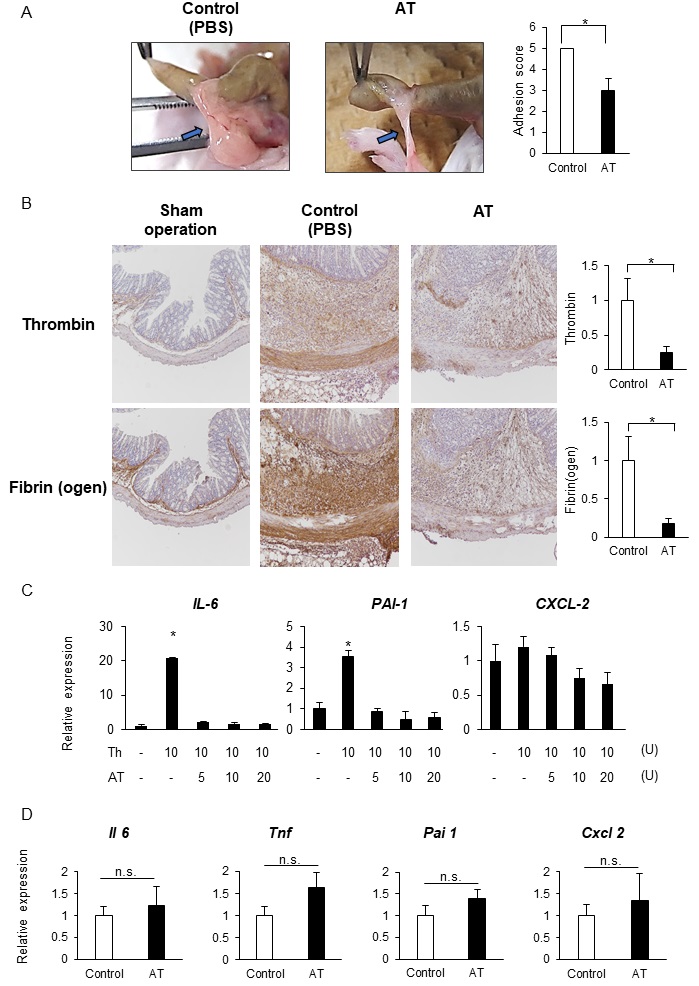
AT failed to reduce neutrophilia or NETs
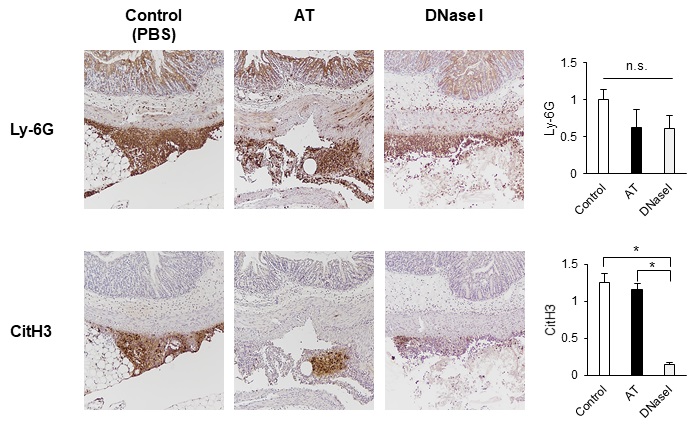
Next, we investigated whether AT treatment affected the migration of neutrophils and/or NET production in the injured serosa. Immunohistochemical analysis for Ly-6G, a surface marker of neutrophils, showed consistent migration of neutrophils into the cauterized serosa in the control mice (Fig. 2, upper left panel and upper graph) and there was no change in the neutrophilia in the AT-treated mice (Fig. 2, upper middle panel and upper graph). This was also consistent with the RT-qPCR analysis which revealed no changes in Cxcl2 (Fig. 1C) expression. NETs were detected using immunohistochemistry against citrullinated histone H3 (CitH3), which is essential for NET release [36]. Mice treated with DNase I, a potent NET inhibitor [37-39], showed a reduction in NET formation without reducing neutrophilia (Fig. 2, right panels and graphs). However, AT-treated mice presented with a similar number of CitH3-positive cells around the cauterization site as the control mice (Fig. 2, lower left and lower middle panels and lower graph), suggesting that AT treatment did not affect postoperative neutrophilia or NET release.
NETs inhibition partly protects against postoperative adhesions
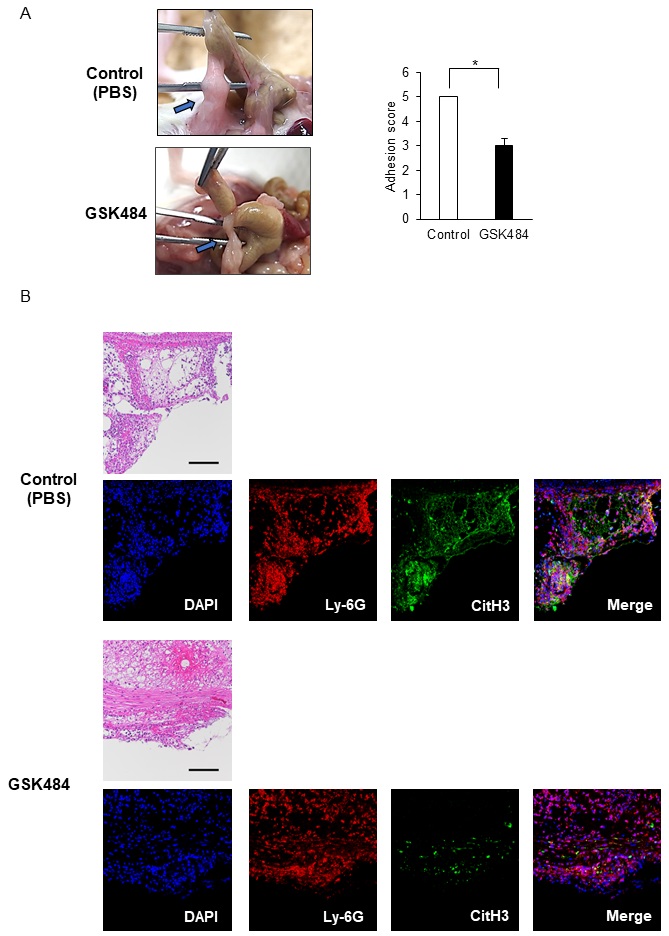
We wanted to know whether NET release is involved in adhesion formation. GSK484 is a selective and reversible inhibitor of peptidyl arginine deiminase (PAD)-4 [40, 41]. Under normal conditions, chromatin is condensed via the acid-base interactions of the arginine residues in the histone proteins. However, in response to the appropriate stimuli neutrophils can induce PAD4 expression which converts these arginine residues into neutral citrulline, leading to chromatin decondensation [36]. This is a key step in NET formation. We administered GSK484 on day1 and 1 h before cecum cauterization. Mice treated with GSK484 presented with a significant but reduced adhesion score compared to the control mice at day 7 post-operation (Fig. 3A). Given this finding we went on to evaluate if the neutrophils are responsible for these NETs, as similar phenotypes can also be produced by several other cell types, including macrophages [10, 42]. This was done using an immunofluorescence assay designed to detect Ly-6G and CitH3. Our evaluations showed no differences in Ly-6G fluorescence intensity between the control and GSK484-treated mice (Fig. 3B). In contrast, GSK484-treated mice displayed much lower intensities of CitH3 than control mice (Fig. 3B) and there were significantly more neutrophils undergoing NETs (stained in yellow) in the control mice than in the GSK484-treatment group (Fig. 3B). Consistent with this, ImageJ Fiji analysis revealed high correlation coefficient of colocalization between Ly-6G and CitH3 in control mice (Supplementary Fig. 2). In contrast, the correlation coefficient was low in the mice receiving GSK484 (Supplementary Fig. 2). These results suggest that GSK484 treatment protects against adhesions, likely through inhibiting NET release by neutrophils.
Combinations of AT and a PAD4 inhibitor abolishes post-operative adhesions
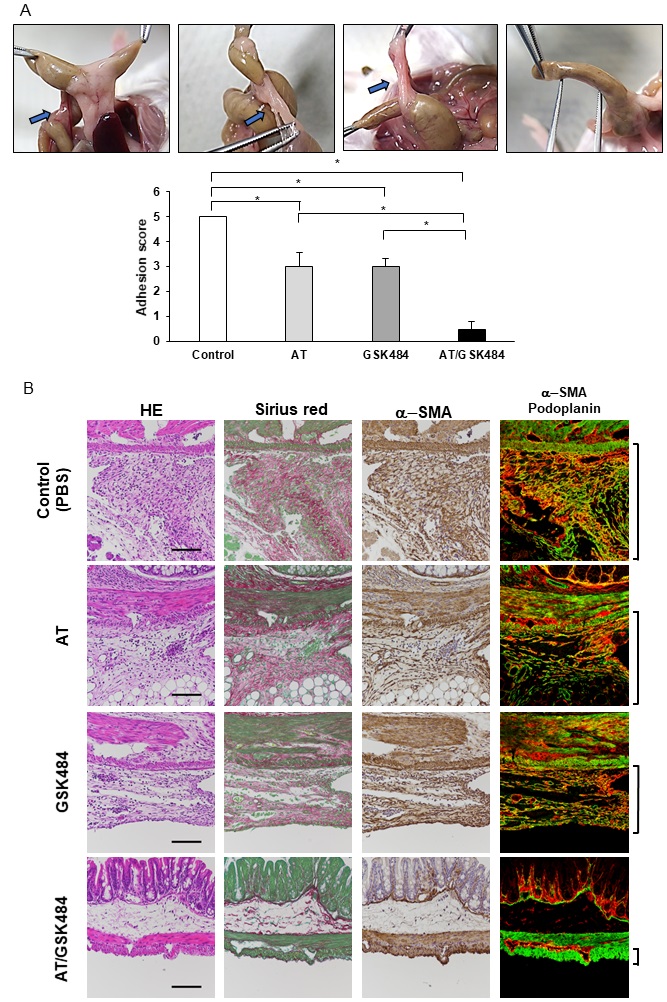
While both AT and GSK484 treatment reduced the severity of the post-operative adhesions neither monotherapy could totally prevent their development (Fig. 1A, 3A). NETs liberate neutrophil-derived proteinases and some of these, such as elastase, have the potential to degrade AT, suggesting that these NETs may reduce its overall efficacy and reduce its therapeutic effect (Fig. 1A). If this is the case, NET inhibition might promote better AT activity and help to further reduce adhesion development. To investigate this, we injected AT in combination with GSK484 into our murine model and compared its combined protective effects to those of AT alone. As expected, these combined therapies facilitated almost complete protection against postoperative adhesions (Fig. 4A). Which was consistent with the aforementioned results (Fig. 1A, 3A), where monotherapy with either AT or GSK484 reduced but did not eliminate the development of post-operative adhesions (Fig. 4A). This was further substantiated by the histological changes in each experimental group (Fig. 4B). The Sirius red-positive fibrosis area within the adhesion band was thinner, but still apparent, in mice that received AT or GSK484 monotherapy when compared to the control, while mice that received both inhibitors demonstrated very few Sirius red-positive areas around the injured serosa (Fig. 4B). We also analyzed myofibroblast development from mesothelial cells using a combination of α-SMA and podoplanin, a cellular marker of mesothelial cells, staining. The numbers of α-SMA+ podoplanin+ cells in mice treated with either inhibitor appeared to be comparable to the control, while α-SMA+ podoplanin+ cells were almost absent in mice treated with both AT and GSK484 (Fig. 4B). In line with this, ImageJ Fiji analysis estimated comparably high correlation coefficients of colocalization between α-SMA and podoplanin in the control, AT-, and GSK484-treated mice (Supplementary Fig. 3). In contrast, the correlation coefficient was low in the mice receiving both AT and GSK484 (Supplementary Fig. 3). These results suggest that postoperative adhesion might be completely prevented by the combined application of both AT and a PAD4 inhibitor.
We thank Dr. Naoki Uyama for helpful discussions. We also thank Yoshimi Miyata for their assistance with the experiments.
Author Contributions
J.F., M.S, and H.T. conceptualized the study; M.S., J. X., K.M., M.J., and J.F. performed experiments; M.S., J. X., H.T., and J.F. analyzed the data; M.S., J. X., H.T., H. E., and J.F. wrote the manuscript; and all authors reviewed and approved the final version of the manuscript.
Funding
This work was supported by the Japan Society for the Promotion of Scientific Research (JSPS) KAKENHI Grant Number JP18H02885.
Statement of Ethics
Animal experiments conform to internationally accepted standards and have been approved by the Institutional Animal Care Committee, Hyogo College of Medicine (approval protocol number: 17-019).
All of the authors have no conflicts of interest.
| 1 Holmdahl L: Making and covering of surgical footprints. Lancet 1999;353:1456-1457. https://doi.org/10.1016/S0140-6736(99)90061-2 |
||||
| 2 Cheong YC, Laird SM, Li TC, Shelton JB, Ledger WL, Cooke ID: Peritoneal healing and adhesion formation/reformation. Hum Reprod Update 2001;7:556-566. https://doi.org/10.1093/humupd/7.6.556 |
||||
| 3 Vrijland WW, Jeekel J, van Geldorp HJ, Swank DJ, Bonjer HJ: Abdominal adhesions: intestinal obstruction, pain, and infertility. Surg Endosc 2003;17:1017-1022. https://doi.org/10.1007/s00464-002-9208-9 |
||||
| 4 ten Broek RP, Issa Y, van Santbrink EJ, Bouvy ND, Kruitwagen RF, Jeekel J, Bakkum EA, Rovers MM, van Goor H: Burden of adhesions in abdominal and pelvic surgery: systematic review and meta-analysis. BMJ 2013;347:f5588. https://doi.org/10.1136/bmj.f5588 |
||||
| 5 Ward BC, Panitch A: Abdominal adhesions: current and novel therapies. J Surg Res 2011;165:91-111. https://doi.org/10.1016/j.jss.2009.09.015 |
||||
| 6 Lin LX, Yuan F, Zhang HH, Liao NN, Luo JW, Sun YL: Evaluation of surgical anti-adhesion products to reduce postsurgical intra-abdominal adhesion formation in a rat model. PLoS One 2017;12:e0172088. https://doi.org/10.1371/journal.pone.0172088 |
||||
| 7 Uyama N, Tsutsui H, Wu S, Yasuda K, Hatano E, Qin XY, Kojima S, Fujimoto J: Anti-interleukin-6 receptor antibody treatment ameliorates postoperative adhesion formation. Sci Rep 2019;9:17558. https://doi.org/10.1038/s41598-019-54175-1 |
||||
| 8 Sudo M, Iida K, Tsutsui H, Mitani K, Jimbo M, Hatano E, Fujimoto J: Blockade of Tumor Necrosis Factor by Etanercept Prevents Postoperative Adhesion Formation in Mice. Cell Physiol Biochem 2020;54:1041-1053. https://doi.org/10.33594/000000286 |
||||
| 9 Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A: Neutrophil extracellular traps kill bacteria. Science 2004;303:1532-1535. https://doi.org/10.1126/science.1092385 |
||||
| 10 Jorch SK, Kubes P: An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 2017;23:279-287. https://doi.org/10.1038/nm.4294 |
||||
| 11 Papayannopoulos V: Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 2018;18:134-147. https://doi.org/10.1038/nri.2017.105 |
||||
| 12 Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD: Neutrophils: New insights and open questions. Sci Immunol 2018;3:eaat4579. https://doi.org/10.1126/sciimmunol.aat4579 |
||||
| 13 Uddin M, Watz H, Malmgren A, Pedersen F: NETopathic Inflammation in Chronic Obstructive Pulmonary Disease and Severe Asthma. Front Immunol 2019;10:47. https://doi.org/10.3389/fimmu.2019.00047 |
||||
| 14 Honda M, Kubes P: Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol 2018;15:206-221. https://doi.org/10.1038/nrgastro.2017.183 |
||||
| 15 Cesarman-Maus G, Hajjar KA: Molecular mechanisms of fibrinolysis. Br J Haematol 2005;129:307-321. https://doi.org/10.1111/j.1365-2141.2005.05444.x |
||||
| 16 Kosaka H, Yoshimoto T, Fujimoto J, Nakanishi K: Interferon-gamma is a therapeutic target molecule for prevention of postoperative adhesion formation. Nat Med 2008;14:437-441. https://doi.org/10.1038/nm1733 |
||||
| 17 Ohashi K, Yoshimoto T, Kosaka H, Hirano T, Iimuro Y, Nakanishi K, Fujimoto J: Interferon γ and plasminogen activator inhibitor 1 regulate adhesion formation after partial hepatectomy. Br J Surg 2014;101:398-407. https://doi.org/10.1002/bjs.9405 |
||||
| 18 Posma JJ, Posthuma JJ, Spronk HM: Coagulation and non-coagulation effects of thrombin. J Thromb Haemost 2016;14:1908-1916. https://doi.org/10.1111/jth.13441 |
||||
| 19 Roemisch J, Gray E, Hoffmann JN, Wiedermann CJ: Antithrombin: a new look at the actions of a serine protease inhibitor. Blood Coagul Fibrinolysis 2002;13:657-670. https://doi.org/10.1097/00001721-200212000-00001 |
||||
| 20 Patnaik MM, Moll S: Inherited antithrombin deficiency: a review. Haemophilia 2008;14:1229-1239. https://doi.org/10.1111/j.1365-2516.2008.01830.x |
||||
| 21 Li W, Johnson DJ, Esmon CT, Huntington JA: Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol 2004;11:857-862. https://doi.org/10.1038/nsmb811 |
||||
| 22 Monroe DM, Hoffman M, Roberts HR: Platelets and thrombin generation. Arterioscler Thromb Vasc Biol 2002;22:1381-1389. https://doi.org/10.1161/01.ATV.0000031340.68494.34 |
||||
| 23 Yeaman MR: Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol 2014;12:426-437. https://doi.org/10.1038/nrmicro3269 |
||||
| 24 Kim SJ, Jenne CN: Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin Immunol 2016;28:546-554. https://doi.org/10.1016/j.smim.2016.10.013 |
||||
| 25 von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, et al.: Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 2012;209:819-835. https://doi.org/10.1084/jem.20112322 |
||||
| 26 Varju I, Kolev K: Networks that stop the flow: A fresh look at fibrin and neutrophil extracellular traps. Thromb Res 2019;182:1-11. https://doi.org/10.1016/j.thromres.2019.08.003 |
||||
| 27 Jordan RE, Nelson RM, Kilpatrick J, Newgren JO, Esmon PC, Fournel MA: Inactivation of human antithrombin by neutrophil elastase. Kinetics of the heparin-dependent reaction. J Biol Chem 1989;264:10493-10500. https://doi.org/10.1016/S0021-9258(18)81648-9 |
||||
| 28 Korkmaz B, Horwitz MS, Jenne DE, Gauthier F: Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010;62:726-759. https://doi.org/10.1124/pr.110.002733 |
||||
| 29 Moser R, Groscurth P, Fehr J: Promotion of transendothelial neutrophil passage by human thrombin. J Cell Sci 1990;96:737-744. https://doi.org/10.1242/jcs.96.4.737 |
||||
| 30 Shankar R, de la Motte CA, Poptic EJ, DiCorleto PE: Thrombin receptor-activating peptides differentially stimulate platelet-derived growth factor production, monocytic cell adhesion, and E-selectin expression in human umbilical vein endothelial cells. J Biol Chem 1994;269:13936-13941. https://doi.org/10.1016/S0021-9258(17)36738-8 |
||||
| 31 Sugama Y, Tiruppathi C, offakidevi K, Andersen TT, Fenton JW, 2nd, Malik AB: Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: a mechanism for stabilizing neutrophil adhesion. J Cell Biol 1992;119:935-944. https://doi.org/10.1083/jcb.119.4.935 |
||||
| 32 Jacques SL, LeMasurier M, Sheridan PJ, Seeley SK, Kuliopulos A: Substrate-assisted catalysis of the PAR1 thrombin receptor. Enhancement of macromolecular association and cleavage. J Biol Chem 2000;275:40671-40678. https://doi.org/10.1074/jbc.M004544200 |
||||
| 33 Coughlin SR: Thrombin signalling and protease-activated receptors. Nature 2000;407:258-264. https://doi.org/10.1038/35025229 |
||||
| 34 Ossovskaya VS, Bunnett NW: Protease-activated receptors: contribution to physiology and disease. Physiol Rev 2004;84:579-621. https://doi.org/10.1152/physrev.00028.2003 |
||||
| 35 Tsai JM, Sinha R, Seita J, Fernhoff N, Christ S, Koopmans T, Krampitz GW, McKenna KM, Xing L, Sandholzer M, Sales JH, Shoham M, McCracken M, Joubert LM, Gordon SR, Poux N, Wernig G, Norton JA, Weissman IL, Rinkevich Y: Surgical adhesions in mice are derived from mesothelial cells and can be targeted by antibodies against mesothelial markers. Sci Transl Med 2018;10:eaan6735. https://doi.org/10.1126/scitranslmed.aan6735 |
||||
| 36 Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA: Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009;184:205-213. https://doi.org/10.1083/jcb.200806072 |
||||
| 37 Macanovic M, Sinicropi D, Shak S, Baughman S, Thiru S, Lachmann PJ: The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin Exp Immunol 1996;106:243-252. https://doi.org/10.1046/j.1365-2249.1996.d01-839.x |
||||
| 38 Jarrot PA, Tellier E, Plantureux L, Crescence L, Robert S, Chareyre C, Daniel L, Secq V, Garcia S, Dignat-George F, Panicot-Dubois L, Dubois C, Kaplanski G: Neutrophil extracellular traps are associated with the pathogenesis of diffuse alveolar hemorrhage in murine lupus. J Autoimmun 2019;100:120-130. https://doi.org/10.1016/j.jaut.2019.03.009 |
||||
| 39 Leffler J, Gullstrand B, Jonsen A, Nilsson JA, Martin M, Blom AM, Bengtsson AA: Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res Ther 2013;15:R84. https://doi.org/10.1186/ar4264 |
||||
| 40 Meng W, Paunel-Gorgulu A, Flohe S, Hoffmann A, Witte I, MacKenzie C, Baldus SE, Windolf J, Logters TT: Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Crit Care 2012;16:R137. https://doi.org/10.1186/cc11442 |
||||
| 41 Cedervall J, Dragomir A, Saupe F, Zhang Y, Arnlov J, Larsson E, Dimberg A, Larsson A, Olsson AK: Pharmacological targeting of peptidylarginine deiminase 4 prevents cancer-associated kidney injury in mice. Oncoimmunology 2017;6:e1320009. https://doi.org/10.1080/2162402X.2017.1320009 |
||||
| 42 Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K, Hase K, Homma K, Sasaki J, Miyauchi H, Hoshino T, Hayashi M, Mayadas TN, Hirahashi J: Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med 2018;24:232-238. https://doi.org/10.1038/nm.4462 |
||||
| 43 Chung JH, Gikakis N, Rao AK, Drake TA, Colman RW, Edmunds LH, Jr.: Pericardial blood activates the extrinsic coagulation pathway during clinical cardiopulmonary bypass. Circulation 1996;93:2014-2018. https://doi.org/10.1161/01.CIR.93.11.2014 |
||||
| 44 Engelmann B, Massberg S: Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 2013;13:34-45. https://doi.org/10.1038/nri3345 |
||||
| 45 Mondal S, Thompson PR: Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc Chem Res 2019;52:818-832. https://doi.org/10.1021/acs.accounts.9b00024 |
||||
| 46 Darrah E, Andrade F: Rheumatoid arthritis and citrullination. Curr Opin Rheumatol 2018;30:72-78. https://doi.org/10.1097/BOR.0000000000000452 |
||||
| 47 Chang X, Yamada R, Sawada T, Suzuki A, Kochi Y, Yamamoto K: The inhibition of antithrombin by peptidylarginine deiminase 4 may contribute to pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2005;44:293-298. https://doi.org/10.1093/rheumatology/keh473 |
||||
| 48 Zarbock A, Polanowska-Grabowska RK, Ley K: Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev 2007;21:99-111. https://doi.org/10.1016/j.blre.2006.06.001 |
||||
| 49 Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR: Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest 1999;103:879-887. https://doi.org/10.1172/JCI6042 |
||||
| 50 Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, Sivridis E, Koffa M, Giatromanolaki A, Boumpas DT, Ritis K, Kambas K: Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol 2014;233:294-307. https://doi.org/10.1002/path.4359 |
||||
| 51 Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM, Wagner DD: Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med 2017;214:439-458. https://doi.org/10.1084/jem.20160530 |
||||