×
![]()
Corresponding Author: Geoffrey W. Abbott
Bioelectricity Laboratory, Department of Physiology and Biophysics, Medical Sciences D,
ZOT 4560, School of Medicine, University of California, Irvine, CA 92697 (USA)
Tel. +1-949 924 3269 , E-Mail abbottg@uci.edu
Rían W. Manvillea Geoffrey W. Abbotta
aBioelectricity Laboratory, Department of Physiology and Biophysics, School of Medicine, University of California, Irvine, CA, USA
Introduction
The Amyloid Precursor Protein (APP) is involved in the regulation of multiple cellular functions via protein-protein interactions and has been most studied with respect to Alzheimer’s disease (AD). Abnormal processing of the C99 fragment of APP contributes to the formation of amyloid plaques, which are causally related to AD. In particular, increases in C99 levels in the brain are thought to contribute to cognitive defects in the early stages of AD. For example, increased C99 levels inside neurons were recently linked to defects in spatial information acquisition, alterations in synaptic plasticity, and early apathy-like behavior in the triple transgenic (3×TgAD: APPswe, TauP301L, PS1M146V) mouse model of AD [1]. Toxic accumulation of C99 is also associated with early anatomical signatures of AD, and may even explain some undesirable side-effects of therapies that inhibit γ-secretase [2]. Importantly, C99 was previously identified as accumulating age-dependently and hippocampus-specifically in 3×TgAD mice, before Aβ accumulation, and with a timing consistent with a role in initiating the neurodegeneration and cognitive dysfunction in 3×TgAD mice [3].
Neuronal Kv channels, including the KCNQ channels that generate the widespread neuronal M-current (predominantly KCNQ2/3 heteromers), are important for learning and memory, and other cognitive functions. De novo KCNQ2 mutations cause early onset epileptic encephalopathies that include seizures but also severe cognitive impairment [4]. KCNQ2 is expressed more highly in human hippocampus than in other brain regions, and KCNQ2 gene variants and expression analysis suggest it is associated with cognitive decline in normal aging [5].
The KCNE family of K+ channel subunits (KCNE1-5) are single transmembrane (TM) domain glycoproteins that are formed from short polypeptides of ~130 amino acids. They co-assemble with a wide range of voltage-gated potassium (Kv) channel pore-forming α subunits, modifying channel functional properties including current density, gating kinetics, voltage dependence, trafficking and pharmacology. KCNE proteins are essential components of many Kv channels in a wide variety of organs and cell types throughout the body [6-8]. Consequently, inherited gene variants in KCNE genes are linked or associated with human diseases of abnormal electrical excitability, including Long QT syndrome, deafness, Brugada syndrome and periodic paralysis [9-13]. Further, Kcne gene deletion in mice causes a plethora of disorders ranging from cardiac arrhythmias [14] and deafness [15, 16] as in human KCNE-linked syndromes, to hypothyroidism [17], gastric cancer [18], atherosclerosis [19], diabetes [20] and increased seizure susceptibility [21].
Given that C99 and KCNEs each exhibit a single transmembrane span topology with an extracellular N-terminal domain and intracellular C-terminal domain, we examined their sequence similarity and the functional effects of C99 on a range of different Kv channel isoforms. Strikingly, C99 is highly effective at modulating Kv channel activity.
Materials and Methods
Channel subunit cRNA preparation and Xenopus laevis oocyte injection
We generated cRNA transcripts encoding human C99, KCNE1, KCNQ1, KCNQ2, KCNQ3, KCNQ4, KCNQ5, Kv1.1, Kv1.2, Kv4.2, Kv4.3S, Kv4.3L and hERG by in vitro transcription using the T7 polymerase mMessage mMachine kit (Thermo Fisher Scientific), after vector linearization, from cDNA sub-cloned into plasmids incorporating Xenopus laevis β-globin 5’ and 3’ UTRs flanking the coding region to enhance translation and cRNA stability. pCAX APP C99 cDNA was a gift from Dennis Selkoe & Tracy Young-Pearse (Addgene plasmid # 30146) [22]. We also generated cRNA from synthesized C99 cDNA (wild-type and with N- and C-terminal flag tag epitopes) and APP in pCDNA3.1+ (codon-optimized and with Xenopus β-globin 5’ and 3’ untranslated regions to enhance RNA stability and translation) (Biomatik, Wilmington, DE) to confirm results with a different source of plasmid and also facilitate biochemical analysis. We quantified cRNA by spectrophotometry. We injected commercially sourced, defolliculated stage V and VI Xenopus laevis oocytes (Ecocyte Bioscience, Austin, TX and Xenoocyte, Dexter, MI) with channel α subunit cRNAs (1-10 ng per oocyte depending on the isoform) with versus without C99 cRNA (5 ng per oocyte) or APP cRNA (7.5 ng/oocyte). We incubated the oocytes at 16 oC in Barth’s saline solution (Ecocyte) containing penicillin and streptomycin, with daily washing, for 2-5 days before two-electrode voltage-clamp (TEVC) recordings.
Two-electrode voltage clamp (TEVC)
We conducted TEVC recordings at room temperature using an OC-725C amplifier (Warner Instruments, Hamden, CT) and pClamp8 software (Molecular Devices, Sunnyvale, CA) 2-5 days after cRNA injection, on oocytes placed in a small-volume oocyte bath (Warner) and viewed with a dissection microscope. We sourced chemicals from Sigma (St. Louis, MO, USA). The bath solution was (in mM): 96 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES (pH 7.6), and pipettes were of 1-2 MΩ resistance when filled with 3 M KCl. We recorded currents in response to pulses between -80 mV and + 40 mV at 20 mV intervals, or a single pulse to +40 mV, from a holding potential of -80 mV, to yield current-voltage relationships, current magnitude, and for quantifying activation rate. We quantified channel deactivation at -80 mV after a single pulse to +40 mV (of varying durations as indicated), from a holding potential of -80 mV. We performed electrophysiology data analysis using Clampfit (Molecular Devices), Origin (OriginLab Corporation, Northampton, MA, USA), and Graphpad Prism software (GraphPad, San Diego, CA, USA); values are stated as mean ± SEM. We plotted peak prepulse currents and raw or normalized tail currents against prepulse voltage and fitted the resultant plots with a single Boltzmann function:
Eq. 1:
g = (A1 – A2)
{1 + exp[(V1/2 – V)/Vs]} + A2
where g is the normalized tail conductance, A1 is the initial value at -∞, A2 is the final value at +∞, V1/2 is the half-maximal voltage of activation and Vs the slope factor. We fitted activation and deactivation kinetics with single exponential functions.
Relative permeability calculations
According to the Goldman-Hodgkin-Katz (GHK) voltage equation:
Eq. 2:
Erev = RT/F ln(PK[K+]o + PNa[Na]o + PCl[Cl]i)
(PK[K+]i + PNa[Na]i + PCl[Cl]o)
Where Erev is the absolute reversal potential and P is permeability. This permits calculation of the relative permeability of each ion if concentrations on either side of the membrane are known. We used a modified version of this equation to determine relative permeability of two ions in a system in which only the extracellular ion concentration was known. Thus, we calculated the relative permeability of Rb+, Cs+, and Na+ compared to K+ ions by plotting the I/V relationships with each extracellular ion (100 mM) and comparing them to the I/V relationship with 100 mM extracellular K+ ion, to yield a change in reversal potential (ΔErev) for each ion compared to that of K+. We then calculated permeability ratios for each ion (X) compared to K+ as:
Eq. 3:
∆Erev = Erev,X – Erev,K = RT/zF lnPx/PK
Chemical structures and silico docking
We plotted and viewed the retigabine chemical structure using Jmol, an open-source Java viewer for chemical structures in 3D: http://jmol.org/. For the in silico ligand docking prediction, we first altered the Xenopus laevis KCNQ1 cryoEM structure [23] to incorporate KCNQ3/KCNQ5 residues known to be important for retigabine and ML-213 binding, and their immediate neighbors, followed by energy minimization, as we previously described [24] using the GROMOS 43B1 force field [25] in DeepView [26]. We then performed unguided docking of retigabine using SwissDock with CHARMM forcefields [27, 28].
Immunofluorescence
Frozen rat sciatic nerve sections were purchased from Zyagen (San Diego, CA), permeabilized (not fixed) in TBS with 0.1% Triton X-100, blocked for 1-2 h with 10% donkey serum, 1% BSA in TBS with 0.1% Triton X-100, then incubated overnight with primary antibodies diluted 1/100 in TBS with 0.1% Triton X-100: rabbit anti-APP (Thermo-Fisher), rabbit anti-C99 (Thermo-Fisher), rabbit anti-Ankyrin G (Santa Cruz Biotechnology), and goat anti-KCNQ2 or KCNQ3 (Santa Cruz Biotechnology). After washing 3 x 5 minutes, sections were incubated for 1-2 h in secondary antibodies (1/200 in TBS) raised in donkey (Invitrogen), before a final wash, mounted with DAPI-containing antifade solution, and then visualized on an Olympus BX51 microscope with Cell-Sens software (Olympus).
Co-immunoprecipitations and western blotting
Xenopus oocytes expressing the various subunit combinations were frozen in batches following TEVC recording and stored at -20 oC until western blotting studies. Batches of 10 oocytes were lysed by suspension and repeated pipetting in a 1 ml pipette tip, in “MiRP buffer”: 150 mM NaCl, 50 mM Tris-HCL (pH 7.4), 20 mM
NaF, 10 mM NaVO4, 1 mM phenylmethylsulfonyl fluoride (Thermo Fisher Scientific, Waltham, MA), 1% Nonidet P-40 (Thermo Fisher Scientific), 1% CHAPS (Sigma, St. Louis, MO, USA), 1% Triton X-100 (Thermo Fisher Scientific), and 1% SDS (Sigma) with protease inhibitor cocktail (Sigma). The suspension was rotated end-over-end at 4 oC for 2 hours, then centrifuged for 10 minutes at 5 x g at 4 oC. Supernatants were decanted (avoiding the pellet and uppermost layer) and used immediately for co-immunoprecipitations or frozen for lysate western blots.
For co-immunoprecipitation (co-IP), oocyte lysates were precleared with protein A agarose beads (ThermoFisher Scientific, Chino, CA, US) and then incubated overnight at 4 oC with one of the following antibodies: goat anti-KCNQ2 (Santa Cruz Biotech), rabbit anti-APP (Thermo-Fisher) or rabbit anti-FLAG (Thermo-Fisher). The following day, antibody-protein complexes were precipitated using protein A beads and separated on 4-12% Bis-Tris gels (Invitrogen), transferred onto PVDF membranes (Bio-Rad) and then western blotted as described below.
Oocyte lysates or co-IP beads were resuspended in LDS gel-loading buffer (Thermo Fisher Scientific) containing 25 mM tris(2-carboxyethyl)phosphine, heated for 10 minutes at 65 oC, vortexed, centrifuged for 3 minutes at 5 x g, and then the supernatants/bead eluents were separated by SDS-PAGE. Proteins were transferred (1 hour at 120 V) to PVDF membranes (BioRad, Hercules, CA, USA). After transfer, PVDF membranes were blocked in PBS (pH 7.6) containing 0.1% Tween-20 (PBST) and 5% dried milk for 1 h at room temperature, washed 3 x 5 minutes in PBST, and then incubated overnight at 4oC with antibodies as follows: 1/100 monoclonal mouse anti-KCNQ2 (Santa Crux Biotech), 1/200 rabbit anti-APP (Thermo-Fisher), 1/500 rabbit anti-FLAG (Thermo-Fisher) in PBST containing 5% (w/v) dried milk. Blots were next washed in PBST (3 x 5 minutes), incubated for 2 h at room temperature with 1/5000 Horseradish peroxidase (HRP)-conjugated IgG secondary antibodies (Bio-Rad or Thermo-Fisher) in PBST, and then washed again in PBST (3 x 5 minutes) before chemiluminescent detection using Luminata Forte chemiluminescence substrate (Millipore, Temecula, CA), and analysis using a Gbox system and Gbox software (Syngene).
Statistical analysis
All values are expressed as mean ± SEM. One-way ANOVA was applied to determine statistical significance. All P-values were two-sided; statistical significance was defined as P < 0.05.
Results
Comparison of C99 and KCNE sequences
C99 and KCNEs share a common 1TM topology, each with extracellular N-terminal domains (Fig. 1A). Kv channel complexes, including those formed from KCNQs, contain 4 α subunits and may also contain 2-4 KCNE β subunits (Fig. 1A, B). C99 and KCNEs exhibit greatest sequence similarity in the transmembrane and intracellular membrane-juxtaposed regions (Fig. 1C). C99 similarity with KCNE1 is comparable to similarity among the KCNEs themselves (Fig. 1D), although the C99 sequence puts it outside the KCNE family (Fig. 1E).
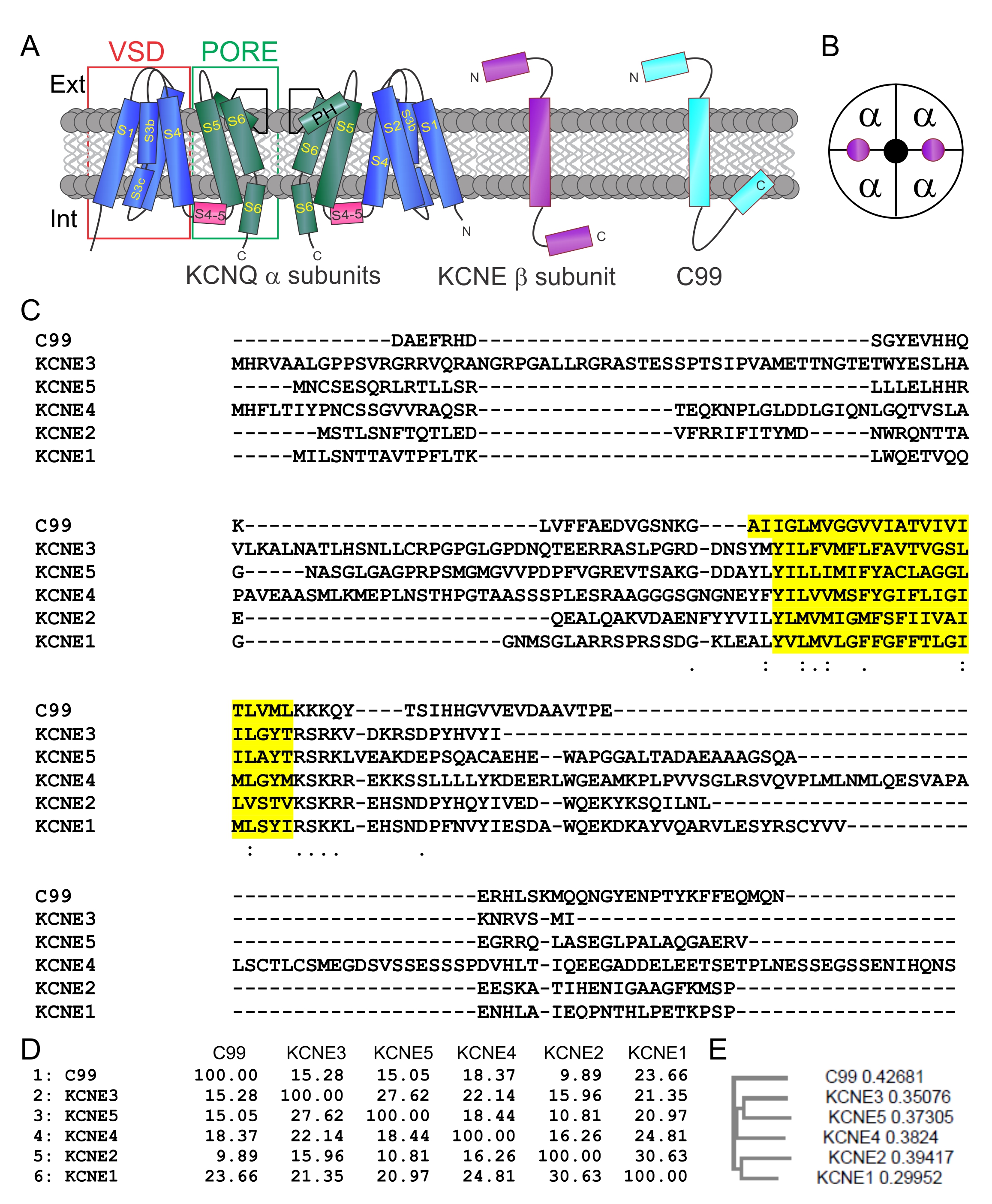
C99 isoform-specifically regulates KCNQ channel function
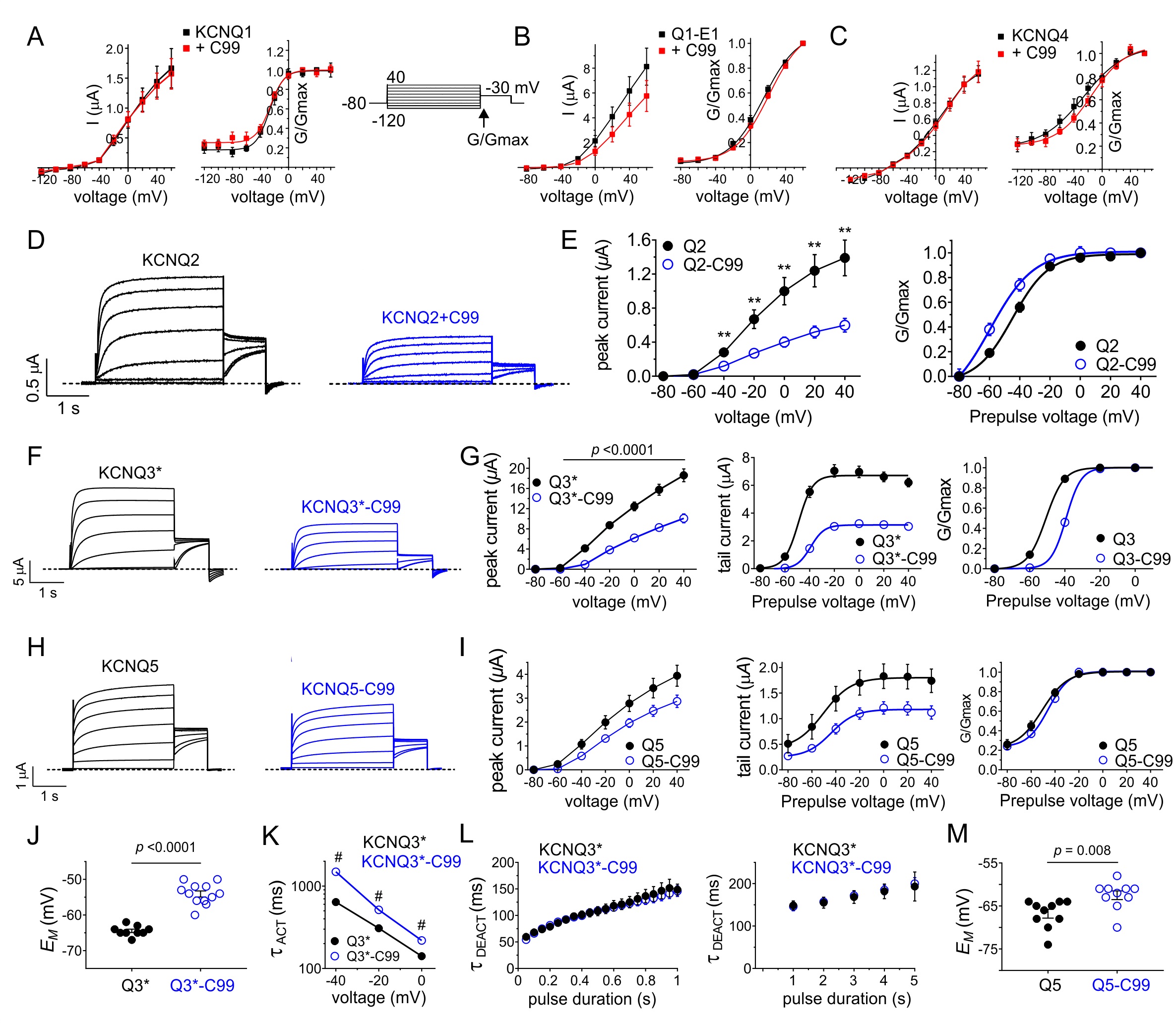
We next tested the functional effects of C99 on KCNQ channels co-expressed in Xenopus laevis oocytes, using TEVC and a voltage family protocol (Fig. 2A upper inset). C99 had no effects on the cardiac and epithelial KCNQ1 channel, in the presence or absence of its cardiac and inner ear β subunit, KCNE1 (Fig. 2A, B). Neither did C99 alter KCNQ4 function (Fig. 2C).
In contrast, C99 exhibited the dual effect of reducing KCNQ2 peak current by >50% and negative-shifted its voltage dependence of activation by -10 mV (Fig. 2D, E). C99 also inhibited KCNQ3* current (a mutant form of KCNQ3 used to ensure robust currents through homomeric KCNQ3 channels [29]), but opposite to effects on KCNQ2, C99 positive-shifted KCNQ3* voltage dependence of activation, >10 mV (Fig. 2F, G). KCNQ5 was less sensitive to C99, which did not shift KCNQ5 voltage dependence of activation (Fig. 2H, I). KCNQ3*-C99 currents were still large enough to analyze in more detail without concerns about contamination from endogenous oocyte currents. Consistent with the positive shift on voltage dependence of KCNQ3* activation, C99 co-expression depolarized the resting membrane potential of oocytes expressing KCNQ3*, compared to those expressing KCNQ3* alone (Fig. 2J). Further, C99 slowed KCNQ3* activation but did not alter deactivation rate, suggesting that C99 destabilized the KCNQ3* open state (Fig. 2K, L). Finally, C99 also slightly depolarized the resting membrane potential of KCNQ5-expressing oocytes, consistent with its subtle effects on KCNQ5 function (Fig. 2M).
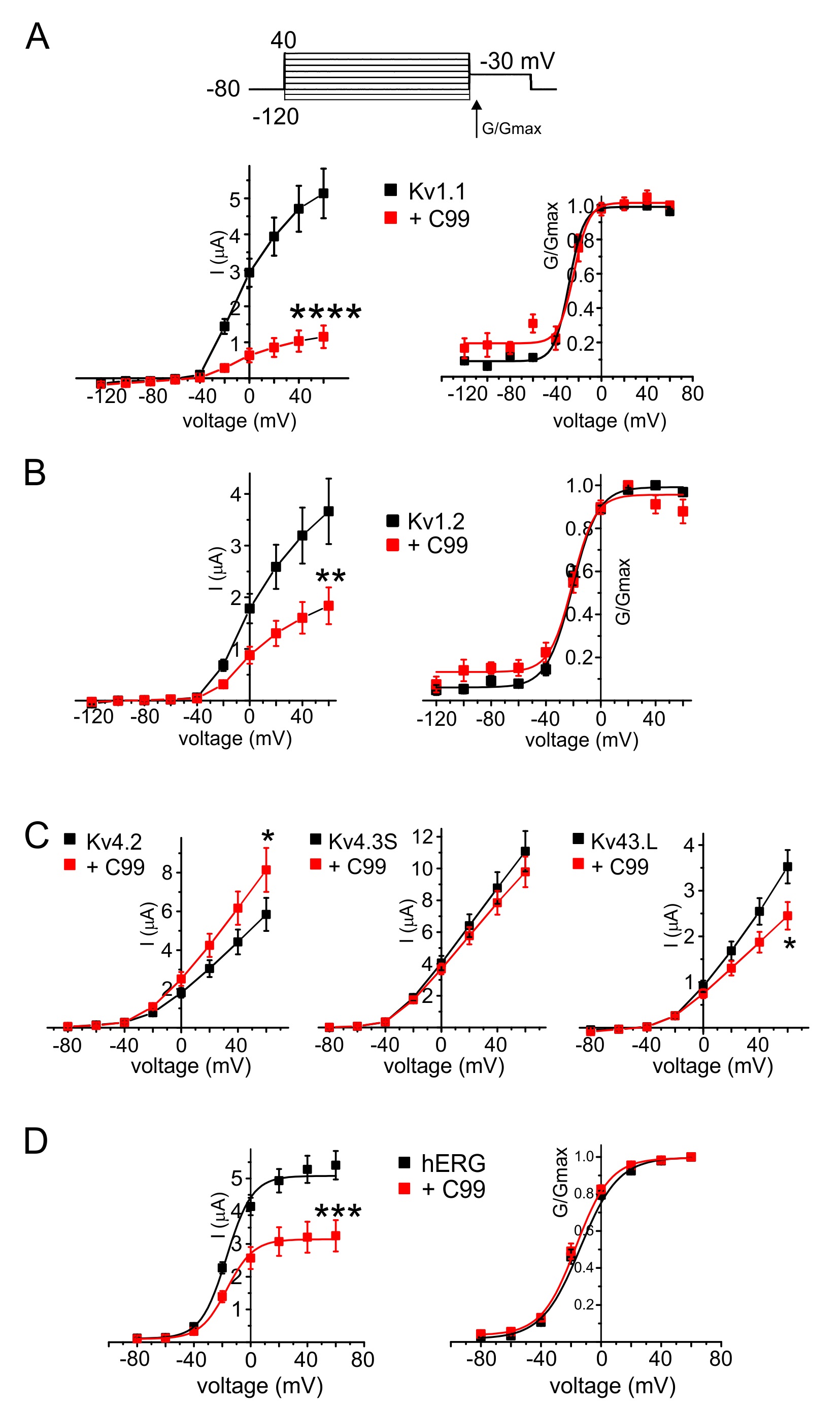
C99 isoform-selectivity inhibits other Kv channels
C99 co-expression also inhibited other Kv channels, outside the KCNQ family. Thus, C99 diminished Kv1.1 (KCNA1) currents by 80%, and Kv1.2 by 50%, without altering the voltage dependence of either (Fig. 3A, B). Effects on Kv4 subfamily channels were varied and relatively small in magnitude: C99 slightly increased Kv4.2 current magnitude and decreased that of Kv4.3L, without changing Kv4.3S current magnitude (Fig. 3C). Finally, C99 decreased hERG current magnitude 40% without altering its voltage dependence (Fig. 3D).
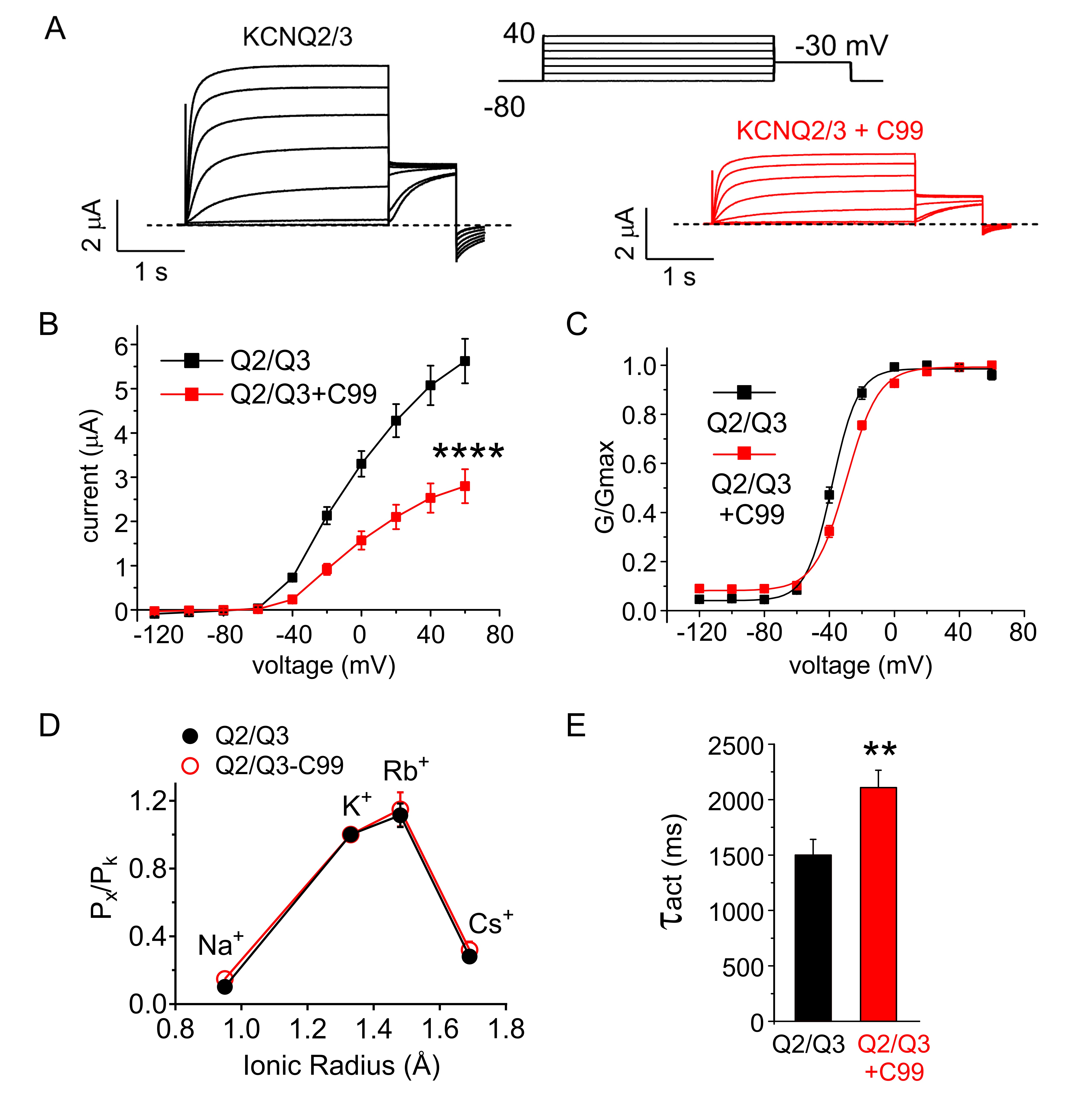
C99 regulates KCNQ2/3 channels
Heteromeric KCNQ2/3 channels are considered to be the predominant molecular correlate of the muscarinic-inhibited neuronal M-current, which is important for controlling neuronal excitability. C99 inhibited KCNQ2/3 peak current by 70%, positive-shifted its voltage dependence of activation and also changed the slope of KCNQ2/3 voltage dependence (Fig. 4A-C). C99 did not alter the ion selectivity of KCNQ2/3 channels (Fig. 4D) but slowed their activation (Fig. 4E).
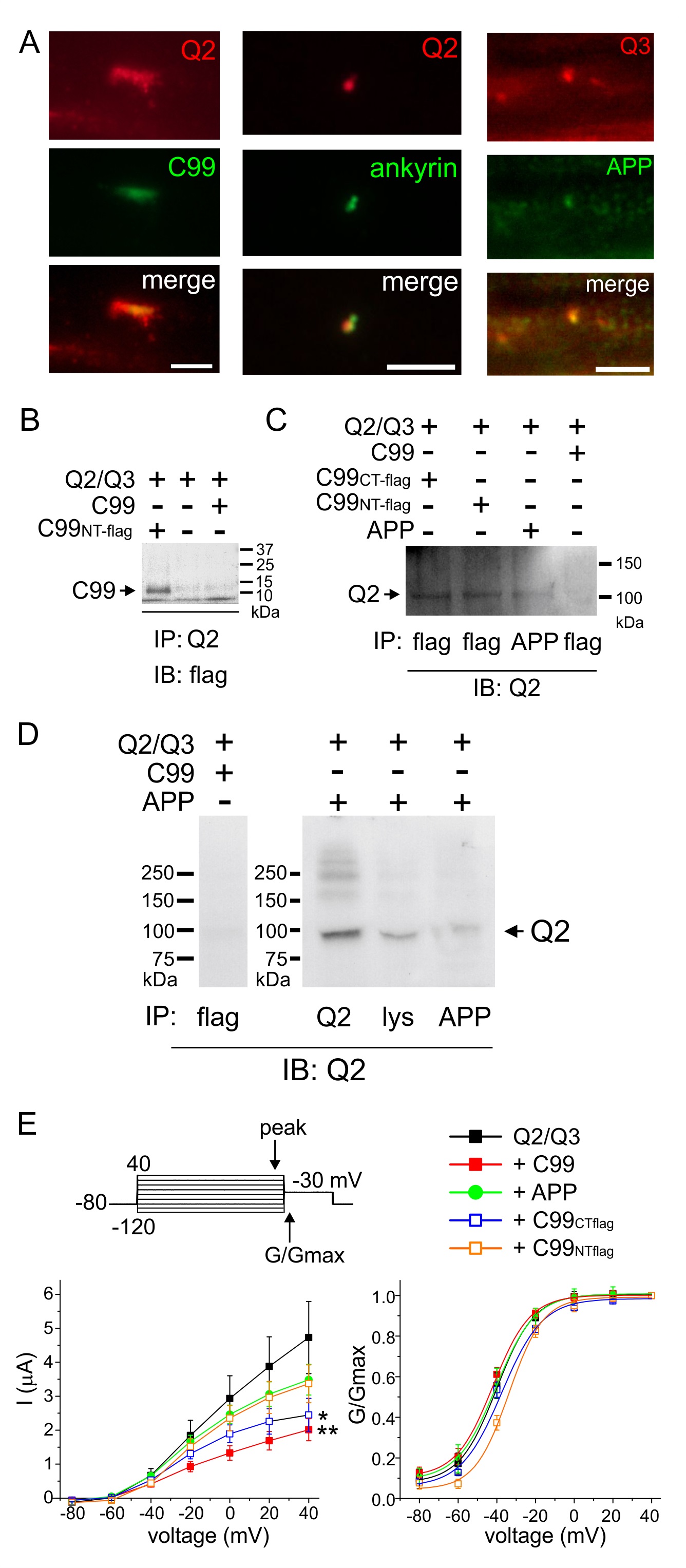
C99 and APP co-localize with KCNQ2/3 in rat sciatic nerve and form complexes in vitro
We and others previously showed that KCNQ2/3 channels are found, among other locations, at the nodes of Ranvier in rat sciatic nerve [30, 31]. Interestingly, others found that APP, the protein that is cleaved to produce C99, also localizes to nodes of Ranvier [32]. Accordingly, here we found that both KCNQ2 and KCNQ3 co-localize in rat sciatic nerve nodes of Ranvier with proteins detected using antibodies raised against C99 and/or APP (Fig. 5A). This suggests that C99 and/or APP regulation of KCNQ2/3 could occur in vivo.
We next generated FLAG-tagged C99 (N- and C-terminal tagged versions) and also wild-type C99 and APP, using synthetic cDNA constructs, to facilitate analysis of complex formation in oocytes. Flag-tagged C99 and wild-type APP each co-immunoprecipitated with KCNQ2 (Fig. 5B-D). As negative controls, untagged C99 was not detected with anti-FLAG antibody (Fig. 5B) and KCNQ2 was not immunoprecipitated with untagged C99 when using an anti-FLAG antibody to immunoprecipitate (Fig. 5C, D).
We also compared the effects of the synthetic construct-generated C99 and APP on KCNQ2/3 function. Synthetic wild-type C99 inhibited KCNQ2/3 by >50%, similar to findings for the original wild-type C99 construct. The FLAG tags partially impaired C99 ability to inhibit KCNQ2/3, possibly indicating effects of the flag tag on either expression or functionality. APP only weakly inhibited KCNQ2/3 function, suggesting cleavage to C99 would increase inhibition within APP-KCNQ2/3 complexes. Effects of any of the proteins on KCNQ2/3 voltage dependence were marginal, as observed for the original wild-type C99 construct (Fig. 5E).
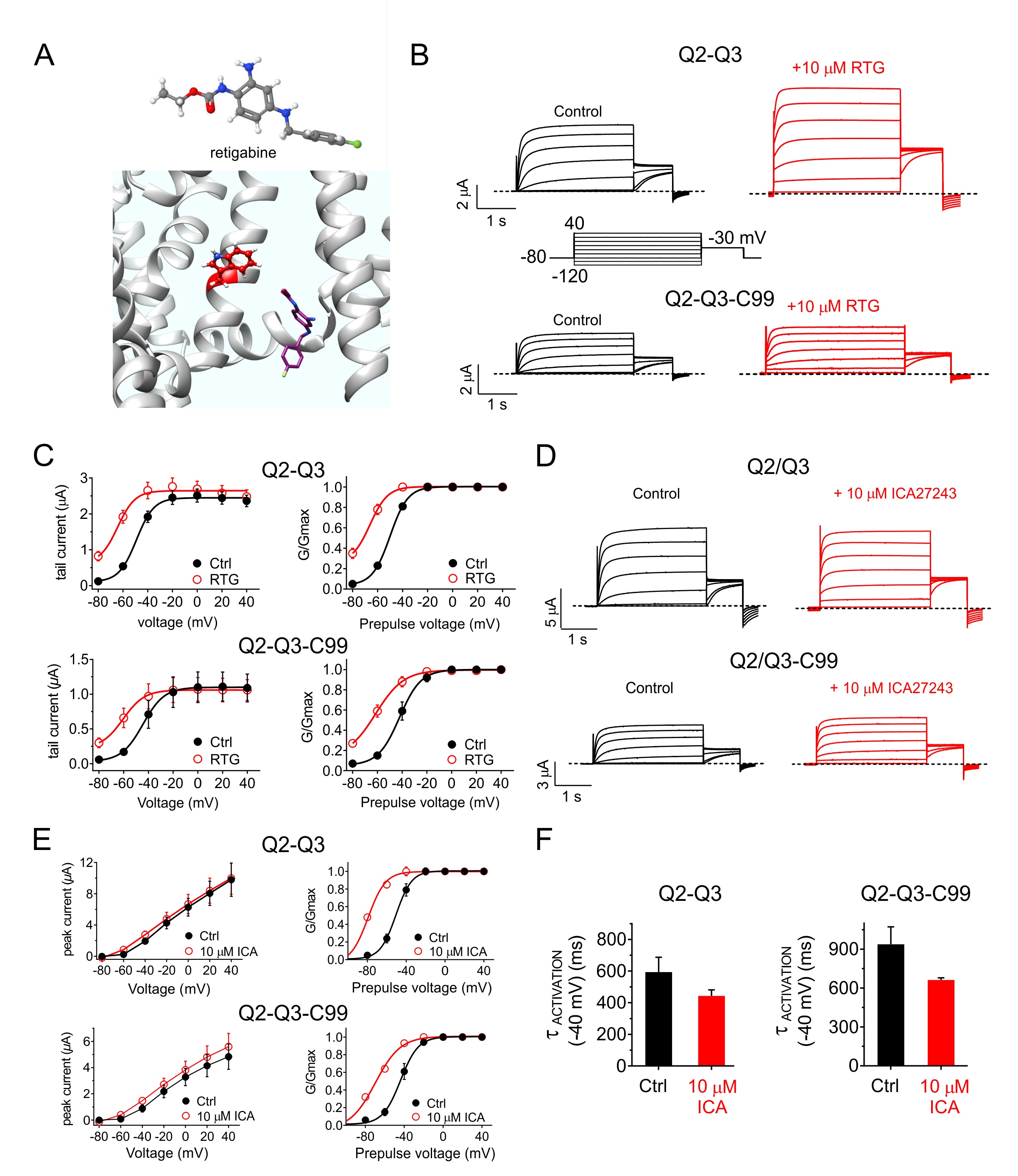
C99 modifies the pharmacological profile of KCNQ2/3 channels
KCNQ2/3 is regulated by a number of small molecules, and new compounds are being specifically developed to open KCNQ2/3 channels with the aim of preventing or treating epilepsy. Retigabine is a KCNQ2/3 opener that requires the S5 residue KCNQ2-W236/KCNQ3-W265 for channel binding and/or activation [33-35] (Fig. 6A). Here, we found that retigabine did not rescue KCNQ2/3 activity from the inhibitory effects of C99. Rather, retigabine exerted similar fold-effects on KCNQ2/3 and KCNQ2/3-C99 channels such that in the presence of drug, C99 was still able to influence KCNQ2/3 activity (Fig. 6B, C). We observed a similar pattern with ICA27243, a KCNQ2/3 opener that is thought to bind to the KCNQ2/3 voltage sensors [36] (Fig. 6D-G).
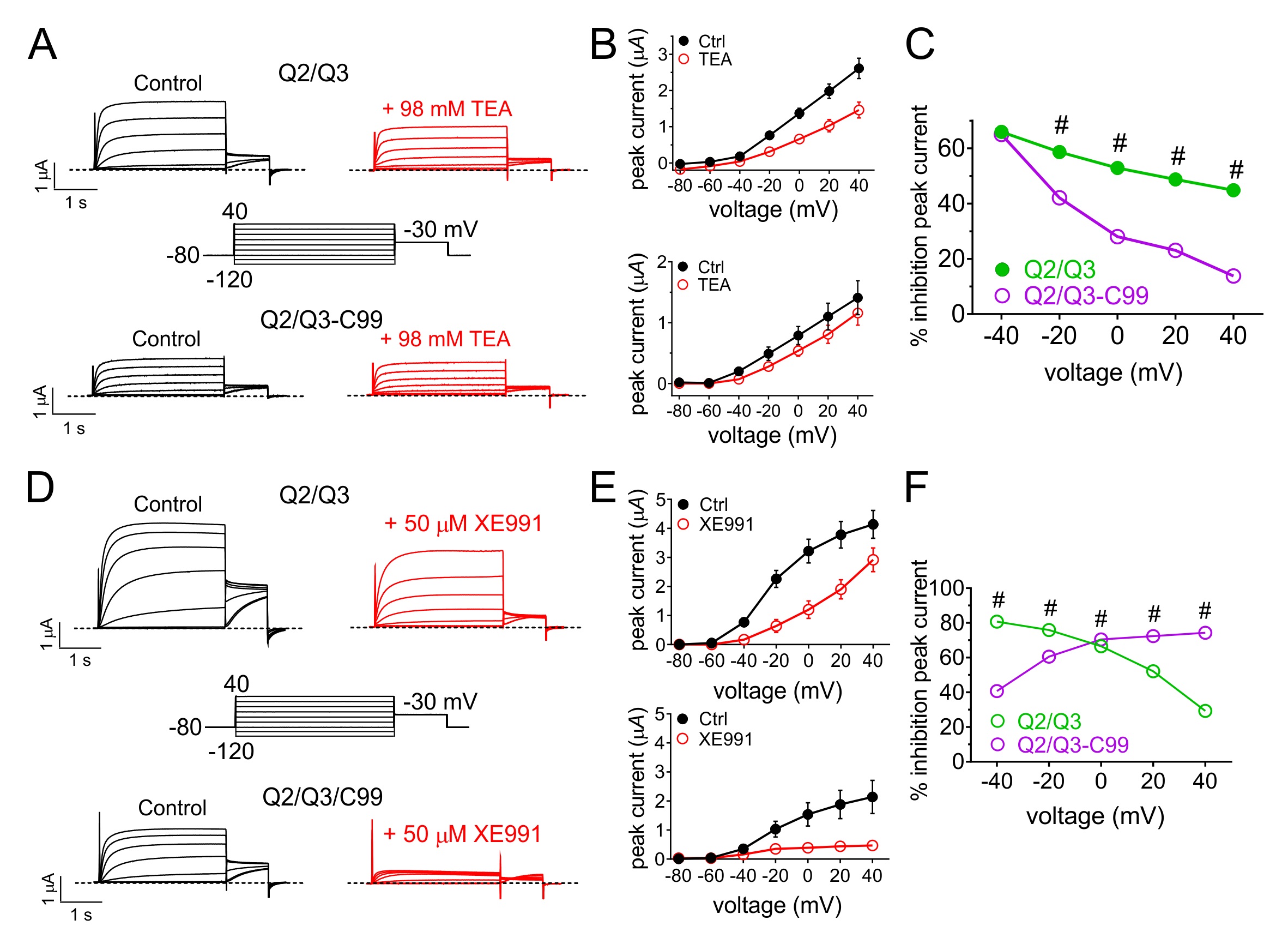
Tetraethylammonium (TEA) is a potassium channel pore blocker that voltage-dependently inhibits many different Kv channels with varying efficacies and potencies [37, 38]. KCNQ2/3 channels are relatively insensitive to TEA but can be blocked by high millimolar amounts. Here, C99 reduced the efficacy of 98 mM TEA on KCNQ2/3, with the most pronounced effects at higher positive voltages (e.g., a threefold reduction in efficacy at +40 mV) (Fig. 7A-C). XE991 is a relatively KCNQ-specific, state-dependent inhibitor thought to bind to a single activated KCNQ2 subunit rather than being an open-channel inhibitor [39]. C99 reversed the voltage dependence of KCNQ2/3 inhibition by XE991, decreasing efficacy at negative potentials but increasing efficacy at positive potentials (e.g., by >twofold at +40 mV (Fig. 7D-F).
Author Contributions
Abbott and Manville performed TEVC experiments and analyses, and prepared figure panels. Abbott conceived the study, performed in silico docking, conducted biochemical experiments, wrote the manuscript and prepared the figures.
Funding
This study was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS107671-02S1 to GWA).
The authors declare that no conflict of interests exists.
| 1 Bourgeois A, Lauritzen I, Lorivel T, Bauer C, Checler F, Pardossi-Piquard R: Intraneuronal accumulation of C99 contributes to synaptic alterations, apathy-like behavior, and spatial learning deficits in 3xTgAD and 2xTgAD mice. Neurobiol Aging 2018;71:21-31. https://doi.org/10.1016/j.neurobiolaging.2018.06.038 |
||||
| 2 Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, Checler F: Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol 2016;132:257-276. https://doi.org/10.1007/s00401-016-1577-6 |
||||
| 3 Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, Fraser P, St-George-Hyslop P, Le Thuc O, Espin V, Chami L, Dunys J, Checler F: The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci 2012;32:16243-16255. https://doi.org/10.1523/JNEUROSCI.2775-12.2012 |
||||
| 4 Weckhuysen S, Mandelstam S, Suls A, Audenaert D, Deconinck T, Claes LR, Deprez L, Smets K, Hristova D, Yordanova I, Jordanova A, Ceulemans B, Jansen A, Hasaerts D, Roelens F, Lagae L, Yendle S, Stanley T, Heron SE, Mulley JC, et al.: KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol 2012;71:15-25. https://doi.org/10.1002/ana.22644 |
||||
| 5 Bonham LW, Evans DS, Liu Y, Cummings SR, Yaffe K, Yokoyama JS: Neurotransmitter Pathway Genes in Cognitive Decline During Aging: Evidence for GNG4 and KCNQ2 Genes. Am J Alzheimers Dis Other Demen 2018;33:153-165. https://doi.org/10.1177/1533317517739384 |
||||
| 6 Abbott GW: The KCNE2 K(+) channel regulatory subunit: Ubiquitous influence, complex pathobiology. Gene 2015;569:162-172. https://doi.org/10.1016/j.gene.2015.06.061 |
||||
| 7 Abbott GW: KCNE1 and KCNE3: The yin and yang of voltage-gated K(+) channel regulation. Gene 2016;576:1-13. https://doi.org/10.1016/j.gene.2015.09.059 |
||||
| 8 Abbott GW: KCNE4 and KCNE5: K(+) channel regulation and cardiac arrhythmogenesis. Gene 2016;593:249-260. https://doi.org/10.1016/j.gene.2016.07.069 |
||||
| 9 Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA: MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 2001;104:217-231. https://doi.org/10.1016/S0092-8674(01)00207-0 |
||||
| 10 Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA: MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999;97:175-187. https://doi.org/10.1016/S0092-8674(00)80728-X |
||||
| 11 Delpon E, Cordeiro JM, Nunez L, Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman-Bang J, Burashnikov E, Christiansen M, Antzelevitch C: Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol 2008;1:209-218. https://doi.org/10.1161/CIRCEP.107.748103 |
||||
| 12 Ohno S, Zankov DP, Ding WG, Itoh H, Makiyama T, Doi T, Shizuta S, Hattori T, Miyamoto A, Naiki N, Hancox JC, Matsuura H, Horie M: KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhyth Electrophysiol 2011;4:352-361. https://doi.org/10.1161/CIRCEP.110.959619 |
||||
| 13 Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT: Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet 1997;17:338-340. https://doi.org/10.1038/ng1197-338 |
||||
| 14 Roepke TK, Kontogeorgis A, Ovanez C, Xu X, Young JB, Purtell K, Goldstein PA, Christini DJ, Peters NS, Akar FG, Gutstein DE, Lerner DJ, Abbott GW: Targeted deletion of kcne2 impairs ventricular repolarization via disruption of I(K,slow1) and I(to,f). FASEB J 2008;22:3648-3660. https://doi.org/10.1096/fj.08-110171 |
||||
| 15 Drici MD, Arrighi I, Chouabe C, Mann JR, Lazdunski M, Romey G, Barhanin J: Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ Res 1998;83:95-102. https://doi.org/10.1161/01.RES.83.1.95 |
||||
| 16 Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J: Inner ear defects induced by null mutation of the isk gene. Neuron 1996;17:1251-1264. https://doi.org/10.1016/S0896-6273(00)80255-X |
||||
| 17 Roepke TK, King EC, Reyna-Neyra A, Paroder M, Purtell K, Koba W, Fine E, Lerner DJ, Carrasco N, Abbott GW: Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med 2009;15:1186-1194. https://doi.org/10.1038/nm.2029 |
||||
| 18 Roepke TK, Purtell K, King EC, La Perle KM, Lerner DJ, Abbott GW: Targeted deletion of Kcne2 causes gastritis cystica profunda and gastric neoplasia. PLoS One 2010;5:e11451. https://doi.org/10.1371/journal.pone.0011451 |
||||
| 19 Lee SM, Nguyen D, Hu Z, Abbott GW: Kcne2 deletion promotes atherosclerosis and diet-dependent sudden death. J Mol Cell Cardiol 2015;87:148-151. https://doi.org/10.1016/j.yjmcc.2015.08.013 |
||||
| 20 Lee SM, Baik J, Nguyen D, Nguyen V, Liu S, Hu Z, Abbott GW: Kcne2 deletion impairs insulin secretion and causes type 2 diabetes mellitus. FASEB J 2017;31:2674-2685. https://doi.org/10.1096/fj.201601347 |
||||
| 21 Abbott GW, Tai KK, Neverisky DL, Hansler A, Hu Z, Roepke TK, Lerner DJ, Chen Q, Liu L, Zupan B, Toth M, Haynes R, Huang X, Demirbas D, Buccafusca R, Gross SS, Kanda VA, Berry GT: KCNQ1, KCNE2, and Na+-Coupled Solute Transporters Form Reciprocally Regulating Complexes That Affect Neuronal Excitability. Sci Signal 2014;7:ra22. https://doi.org/10.1126/scisignal.2005025 |
||||
| 22 Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ: A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci 2007;27:14459-14469. https://doi.org/10.1523/JNEUROSCI.4701-07.2007 |
||||
| 23 Sun J, MacKinnon R: Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 2017;169:1042-1050 e1049. https://doi.org/10.1016/j.cell.2017.05.019 |
||||
| 24 Manville RW, Papanikolaou M, Abbott GW: Direct neurotransmitter activation of voltage-gated potassium channels. Nat Commun 2018;9:1847. https://doi.org/10.1038/s41467-018-04266-w |
||||
| 25 van Gunsteren WF: Biomolecular simulation: the GROMOS96 manual and user guide.. Zürich, Vdf Hochschulverlag an der ETH Zürich, 1996. | ||||
| 26 Johansson MU, Zoete V, Michielin O, Guex N: Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinformatics 2012;13:173. https://doi.org/10.1186/1471-2105-13-173 |
||||
| 27 Grosdidier A, Zoete V, Michielin O: SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res 2011;39:W270-277. https://doi.org/10.1093/nar/gkr366 |
||||
| 28 Grosdidier A, Zoete V, Michielin O: Fast docking using the CHARMM force field with EADock DSS. J Comput Chem 2011;32:2149-2159. https://doi.org/10.1002/jcc.21797 |
||||
| 29 Etxeberria A, Santana-Castro I, Regalado MP, Aivar P, Villarroel A: Three mechanisms underlie KCNQ2/3 heteromeric potassium M-channel potentiation. J Neurosci 2004;24:9146-9152. https://doi.org/10.1523/JNEUROSCI.3194-04.2004 |
||||
| 30 Devaux JJ, Kleopa KA, Cooper EC, Scherer SS: KCNQ2 is a nodal K+ channel. J Neurosci 2004;24:1236-1244. https://doi.org/10.1523/JNEUROSCI.4512-03.2004 |
||||
| 31 Neverisky DL, Abbott GW: KCNQ-SMIT complex formation facilitates ion channel-solute transporter cross talk. FASEB J 2017;31:2828-2838. https://doi.org/10.1096/fj.201601334R |
||||
| 32 Li S, Wang X, Ma QH, Yang WL, Zhang XG, Dawe GS, Xiao ZC: Amyloid precursor protein modulates Nav1.6 sodium channel currents through a Go-coupled JNK pathway. Sci Rep 2016;6:39320. https://doi.org/10.1038/srep39320 |
||||
| 33 Kim RY, Yau MC, Galpin JD, Seebohm G, Ahern CA, Pless SA, Kurata HT: Atomic basis for therapeutic activation of neuronal potassium channels. Nat Commun 2015;6:8116. https://doi.org/10.1038/ncomms9116 |
||||
| 34 Lange W, Geissendorfer J, Schenzer A, Grotzinger J, Seebohm G, Friedrich T, Schwake M: Refinement of the binding site and mode of action of the anticonvulsant Retigabine on KCNQ K+ channels. Mol Pharmacol 2009;75:272-280. https://doi.org/10.1124/mol.108.052282 |
||||
| 35 Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, Schwake M: Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci 2005;25:5051-5060. https://doi.org/10.1523/JNEUROSCI.0128-05.2005 |
||||
| 36 Padilla K, Wickenden AD, Gerlach AC, McCormack K: The KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain site. Neurosci Lett 2009;465:138-142. https://doi.org/10.1016/j.neulet.2009.08.071 |
||||
| 37 Armstrong CM: Time course of TEA(+)-induced anomalous rectification in squid giant axons. J Gen Physiol 1966;50:491-503. https://doi.org/10.1085/jgp.50.2.491 |
||||
| 38 Armstrong CM, Hille B: The inner quaternary ammonium ion receptor in potassium channels of the node of Ranvier. J Gen Physiol 1972;59:388-400. https://doi.org/10.1085/jgp.59.4.388 |
||||
| 39 Greene DL, Kang S, Hoshi N: XE991 and Linopirdine Are State-Dependent Inhibitors for Kv7/KCNQ Channels that Favor Activated Single Subunits. J Pharmacol Exp Ther 2017;362:177-185. https://doi.org/10.1124/jpet.117.241679 |
||||
| 40 Park KH, Kwok SM, Sharon C, Baerga R, Sesti F: N-Glycosylation-dependent block is a novel mechanism for drug-induced cardiac arrhythmia. FASEB J 2003;17:2308-2309. https://doi.org/10.1096/fj.03-0577fje |
||||
| 41 Xu DE, Zhang WM, Yang ZZ, Zhu HM, Yan K, Li S, Bagnard D, Dawe GS, Ma QH, Xiao ZC: Amyloid precursor protein at node of Ranvier modulates nodal formation. Cell Adh Migr 2014;8:396-403. https://doi.org/10.4161/cam.28802 |
||||
| 42 Puig KL, Combs CK: Expression and function of APP and its metabolites outside the central nervous system. Exp Gerontol 2013;48:608-611. https://doi.org/10.1016/j.exger.2012.07.009 |
||||