×
![]()
Corresponding Author: Leonardo F. Ferreira
Department of Applied Physiology and Kinesiology, University of Florida
1864 Stadium Rd, Room 100 FLG, Gainesville, FL 32611-8205 (USA)
Tel. +1-352-294-1724, E-Mail ferreira@hhp.ufl.edu
Nox4 Knockout Does Not Prevent Diaphragm Atrophy, Contractile Dysfunction, or Mitochondrial Maladaptation in the Early Phase Post-Myocardial Infarction in Mice
Dongwoo Hahna Ravi A. Kumara Derek R. Muscatoa Terence E. Ryana Katrin Schröderb Leonardo F. Ferreiraa
aDepartment of Applied Physiology and Kinesiology, College of Health and Human Performance, University of Florida, Gainesville, FL, USA bInstitute for Cardiovascular Physiology, Goethe University, Frankfurt, Germany
Introduction
Myocardial infarction (MI) is one of the most common forms of pathology in the heart and results in cardiomyocyte dysfunction and death due to ischemia [1]. The primary concerns in the care of patients in the early phase post-MI are cardiac revascularization and arrhythmias. However, recent studies in patients and rodents identified that the early phase post-MI is accompanied by inspiratory muscle (diaphragm) weakness and endurance [2-4], which bears relevance to the overall cardiopulmonary pathophysiology [5]. Diaphragm abnormalities contribute to dyspnea [6], impaired airway clearance [7], and elevated sympathetic nervous activity [8] – the latter is a critical determinant of arrhythmias post-MI [9, 10] that can lead to cardiac arrest and death [8, 11].
The early phase post-MI is characterized by elevated circulating inflammatory cytokines and angiotensin II [12, 13], which signal via reactive oxygen species (ROS) [3, 14]. Excess ROS causes muscle weakness due to atrophy and contractile dysfunction [15, 16] and impairs mitochondrial function [17]. A potential source of ROS downstream of inflammatory cytokines and angiotensin II is NADPH oxidase 4 (Nox4) [18, 19]. Nox4, which localizes in the sarcoplasmic reticulum and mitochondria [20-22], causes skeletal muscle atrophy induced by angiotensin II infusion and pancreatic cancer [14, 23], mediates contractile dysfunction in metastatic bone cancer [24], and promotes mitochondrial dysfunction [22]. In the early phase post-MI, the systemic environment is primed to activate Nox4 signaling [23, 25, 26]. However, the role of Nox4 on diaphragm atrophy, contractile dysfunction, and potential mitochondrial abnormalities in the early phase post-MI is unknown.
The main goal of this study was to test the role of Nox4 on diaphragm weakness in the early phase post-MI. Based on the localization of Nox4 in mitochondria [21] and the relevance of mitochondrial function to muscle health in several conditions [27], we aimed to define the impact of MI on diaphragm mitochondrial respiration and ROS emission and the role of Nox4 therein.
Materials and Methods
Animals and ethical approval
We conducted experiments on 7-9 months old male mice: twelve C57BL/6J (WT; Jackson Laboratories) and thirteen whole-body Nox4 knockout (Nox4KO; a gift from Drs. Ralf Brandes and Katrin Schröder). Nox4 knockout mice were generated by targeted deletion of the translation initiation site and exons 1 and 2 of the gene as described previously [28]. We backcrossed the original litter of Nox4KO with C57BL/6J for over ten generations and confirmed the genotype of all Nox4KO mice used in this study (Supplementary Fig. S1 – for all supplementary material see www.cellphysiolbiochem.com) following a previous protocol [29]. We housed the mice at the University of Florida Animal Care Facilities in a 12:12-h light-dark cycle and supplied standard chow and water ad libitum. We followed guidelines set by the National Institutes of Health for all animal procedures performed in this study. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Florida.
Coronary artery ligation
Animals went through ligation of the coronary artery to cause myocardial infarction using aseptic procedures or sham surgery. We anesthetized the animals with a mixture of isoflurane (3-5% for induction, 2-3% for maintenance) and oxygen, shaved the left side of the thorax, and cleaned the surgical area with 4% chlorhexidine gluconate and sterile saline. When animals reached the surgical plane of anesthesia, we performed orotracheal intubation and connected the animal to a rodent respirator (Model 683, Harvard Apparatus Inc. Holliston, MA). We made incisions through the skin using a scalpel blade and bluntly dissected the thoracic muscle layer and intercostal muscles to expose the heart. Once the heart was exposed, we removed the pericardium and ligated the left anterior descending coronary artery close to the left atrium with a 6-0 monofilament absorbable PGA suture (DemeSORB™, DemeTECH, Miami Lakes, FL). After the ligation, we hyperinflated the lungs and closed the chest wall (6-0 PGA suture, DemeSORB™, DemeTECH) and skin (3-0 Nylon, Demelon™, DemeTECH). The animals in the sham group went through the same procedure except for the coronary artery ligation. We injected buprenorphine SR-LAB (ZooPharm, Laramie, WY) subcutaneously (1.0 mg/kg) immediately before surgery for analgesia. We supplied moist chow at the bottom of the cage and kept the cage for three days on top of a water-heated pad (Stryker, Kalamazoo, MI) to maintain the temperature of the cage at ~32°C.
Terminal experiments
We performed terminal experiments 72 hrs post-surgery. After we anesthetized the animal with isoflurane (5% induction, 2-3% maintenance), we performed laparotomy to collect the whole diaphragm and heart. We used the left hemidiaphragm to assess contractile function and the right hemidiaphragm for mitochondrial assays with saponin-permeabilized muscle fiber bundles. The unused portions of the diaphragm were snap-frozen in liquid nitrogen or embedded in Tissue-Tek OCT freezing medium (Sakura Finetek, Torrance, CA), frozen in liquid nitrogen-cooled isopentane, and stored at -80°C. We measured the weight of the heart after removing the atria and saved it in Tissue-Tek OCT freezing medium following the procedures described above for the diaphragm.
Histology
We used a cryostat (CM 3050S, Leica, Biosystems, Buffalo Grove, IL) set at -20°C to collect 10 µm cross-sections of the diaphragm and heart samples frozen in Tissue-Tek OCT freezing medium (Sakura Finetek, Torrance, CA). After transferring sections to the microscope slides, we covered the sections with wheat germ agglutinin (WGA) Texas Red (Molecular Probes, Eugene, OR) diluted in 1:200 at room temperature (15 min for heart, 1 hr for diaphragm). We washed the sections in phosphate-buffered saline (PBS) for
3 × 5 min, allowed them to dry, and followed different approaches for heart and diaphragm sections. For heart sections, we made panoramic images using an inverted microscope (Axio Observer, Zeiss, Thornwood, NY) connected to a camera (AxioCam ERc5s) and Zen Pro software (Zeiss Thornwood, NY). The percentage infarct size was the average percentage of endocardial and epicardial infarct length. We used this approach as WGA lectin binds to N-acetylglycosamin, which is rich in the extracellular matrix and connective tissue in the scar of the infarcted area [30].
For the diaphragm, we permeabilized the sample (after WGA staining and washes) with 0.5% Triton X-100 solution for 5 min, washed in PBS for 5 min, and incubated in primary antibodies in a humid chamber for 1.5 hrs. We used primary antibodies for myosin heavy chain type I (A4.840, 1:15; Developmental Studies Hybridoma Bank, Iowa City, IA) and type IIa (SC-71, 1:50, Developmental Studies Hybridoma Bank, Iowa City, IA). Subsequently, we washed the sample in PBS for 3 × 5 min and incubated it with fluorescently conjugated secondary antibodies (Goat × Mouse IgM Alexa 350 and Goat × Mouse IgG Alexa 488, Invitrogen, Carlsbad, CA) for 1 hr. We washed the sectioned tissue in PBS for 3 × 5 min and allowed it to dry. Lastly, we imaged the sections using an inverted fluorescence microscope (Axio Observer, Zeiss, Thornwood, NY) connected to a fluorescence camera (AxioCam MRm, Zeiss, Thornwood, NY) and Zen Pro software (Zeiss, Thornwood, NY). We used an automated image quantification platform to analyze fiber type distribution and cross-sectional area (MyoVision, University of Kentucky College of Health Sciences, Lexington, KY) [31].
qPCR
We retrieved diaphragm samples from Tissue-Tek OCT freezing medium (Sakura Finetek, Torrance, CA) in sterile PBS and homogenized them in TRI-Reagent (Sigma Aldrich, St. Louis, MO) using stainless steel beads and a bullet blender (Next Advanced, Troy, NY). We isolated RNA by using the Direct-Zol RNA Microprep kit (Zymo Research, Irvine, CA). We assessed RNA quantity and quality with UV spectroscopy (Thermo Fisher, Waltham, MA), then generated cDNA using the Quantabio qScript cDNA synthesis kit (Quantabio, Beverly, MA). Real-time PCR was performed on a Quantstudio 3 thermocycler (Thermo Fisher, Waltham, MA) using Taqman Universal Master Mix II and Taqman probes (all from Thermo Fisher, Waltham, MA) for Nox4 (Mm00479246_m1), Fbxo32 (Mm00499523_m1; MAFbx/atrogin-1), and Trim63 (Mm01185221_m1; MuRF-1). Results were normalized to HPRT (Mm00446968_m1) and gene expression was calculated relative to the WT-Sham group using the ΔΔCT method.
Diaphragm contractile function in vitro
We immersed the left hemidiaphragm in bicarbonate buffered solution (in mM: 137 NaCl, 5 KCl,
1 MgSO4, 1 NaH2PO4, 24 NaHCO3, 2 CaCl2) bubbled with a mixture of 95% O2 and 5% CO2 gas at room temperature and isolated a strip with ribs and central tendon attached to it. We tied the rib to a glass rod (158816; Radnoti, Monrovia, CA) and attached the central tendon to a Dual-Mode Muscle Lever System (300C-LR, Aurora Scientific Inc., Aurora, Canada) with 4-0 silk suture (Fine Scientific, Foster City, CA). We kept the bundle between platinum electrodes connected to a biphasic high-power stimulator (701C, Aurora Scientific Inc., Aurora, Canada) in a water-jacketed organ bath filled with Krebs buffer continuously bubbled with 95% O2 and 5% CO2 gas. We found the optimal length of the bundle (L0) by measuring tetanic force in response to 120 Hz stimulation (600 mA current, 0.25 ms pulse) at room temperature. After we placed the bundle at L0, we increased the temperature of the organ bath and allowed 10 min for thermo-equilibration at 37°C, and stimulated at frequencies of 1, 30, 50, and 300 Hz.
After the protocol, we retrieved the diaphragm bundle from the apparatus, blotted it dry, and measured the bundle weight. We calculated the diaphragm bundle cross-sectional area (CSA, cm2) based on the wet weight (g) and estimated muscle density (1.056 g/cm3) [32] to determine the specific force (N/cm2). We used the ‘high throughput’ function of DMA software (Aurora Scientific, Aurora, Canada) to analyze isometric contractile properties.
Muscle bundle preparation for mitochondrial function assessment
Our optimized approach to prep the mouse diaphragm for saponin-permeabilization of fiber bundles has been described in detail recently [33]. Briefly, we placed the diaphragm sample in fresh ice-cold buffer X (in mM: 7.23 K2EGTA, 2.77 Ca-K2EGTA, 20 imidazole, 20 taurine, 5.7 ATP, 14.3 PCr, 6.56 MgCl2-6H2O, 50 K-MES; pH 7.1) in a tissue culture dish with silicone gel. After removing the ribs, blood clots, adipose and connective tissues, we cut the diaphragm perpendicular to the fiber orientation along the phrenic nerve and peeled off the abdominal fascia and muscle fiber layer. We used the ‘pleural layer’ of diaphragm fibers for bundle preparation, as this side contains a thinner fascia and less connective tissue than the abdominal side [34]. We teased individual fibers longitudinally and left 0.1-0.2 mm of the fibers attached at the end of the bundle to prevent losing fibers during washing and transfer between containers. We trimmed the distal end of the separated fibers to remove portions potentially damaged during the teasing process. We transferred the separated bundles to microcentrifuge tubes with 1.5 ml of buffer X + saponin (30 µg/ml), and rotated the tubes for 30 min at 4°C. Lastly, we washed the bundles for 3 × 5 min at 4°C in a microcentrifuge tube with 1.5 ml buffer Z (in mM: 30 KCl, 10 KH2PO4, 5 MgCl2-6H2O, 105 K-MES, and 0.5 mg/ml BSA; pH 7.1). The individual bundles were then allocated to measurements of mitochondrial respiration (2 bundles) and ROS emission (1 bundle).
Mitochondrial respiration
We measured O2 consumption at 37°C in buffer Z containing 20 mM creatine monohydrate and 10 µM blebbistatin [35] in a high-resolution respirometer (O2K Oxygraph; Oroboros, Innsbruck, Austria). We used Hamilton Syringes (Hamilton, Reno, NV) to add substrates and cytochrome c in the following order: 5 mM pyruvate + 0.5 mM malate (State 2 respiration, complex I), ADP (100 µM, 500 µM; State 3 respiration, complex I), 10 mM cytochrome c (mitochondrial membrane integrity), and 10 mM succinate (State 3 respiration, complex I+II). We initiated the experiment when the O2 concentration of the buffer was stabilized at approximately 400 µM, and reoxygenated the buffer when O2 concentration was less than 250 µM during the protocol. Once we finished the measurements, we retrieved the bundles to quantify the total protein content of the bundles (DC Assay, Bio-Rad Laboratories, Hercules, CA). We completed the experiments in two bundles per mouse and used the average JO2 for data analysis. We excluded bundles that displayed a JO2 increase ≥ 10% upon addition of cytochrome c from analysis (2 bundles from separate animals within WT-MI, 1 bundle from Nox4KO-MI). The bundles used for mitochondrial respiration were retrieved from the O2K chamber, blotted dry, transferred to a microcentrifuge tube, snap-frozen in liquid nitrogen, and stored at -80°C. For measurement of total protein content in permeabilized bundles, we added 50 µl of mitochondrial isolation medium to the microcentrifuge tube, homogenized the samples in the microcentrifuge tube using a motor-driven polypropylene micropestle, centrifuged the homogenate at 500 g for 7 min at 4°C (#5424 Eppendorf, Hauppauge, NY), and used the supernatant for measurement of total protein content (DC Assay, Bio-Rad Laboratories, Hercules, CA). The JO2 data were normalized to total protein per bundle and citrate synthase abundance to estimate intrinsic mitochondrial respiratory function [36].
Mitochondrial reactive oxygen species emission
We used an assay based on Amplex Ultra Red (Life Technologies, Eugene, OR) fluorescence (λEm = 565 nm, λExc = 600 nm) to measure H2O2 emission from permeabilized diaphragm fiber bundles in a fluorometer (Fluorolog-3, Horiba Jobin Yvon Inc., Edison, NJ) with a 1 ml quartz cuvette (14-958-109, Fisherbrand, Pittsburgh, PA) maintained at 37°C. The assay solution contained 10 µM Amplex Ultra Red, 25 µM blebbistatin, 20 mM creatine monohydrate, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2-6H2O, 8 µM BSA, 105 mM K-MES, 1 mM EGTA; pH 7.1, 30 U/ml superoxide dismutase, and 1 U/ml horseradish peroxidase. Initially, we allowed a 5-min period for thermal equilibration and stabilization of the fluorescence signal. Subsequently, we measured H2O2 emission rates from the permeabilized fiber bundles at baseline and after stepwise addition of substrates as in the mitochondrial respiration experiment. Once we finished the measurements, we retrieved the bundle to determine the total protein content of the bundles as described for mitochondrial respiration. Before each round of the experiment, we defined a standard curve for the relationship between Amplex Ultra Red fluorescence and [H2O2], converted fluorescence values to [H2O2], and subtracted baseline H2O2 rate to obtain the substrate-induced H2O2 flux, JH2O2. We calculated the JH2O2/JO2 × 100 under matching conditions of JH2O2 and JO2 measurements to determine the percentage mitochondrial ETS electron leak.
SDS-PAGE and immunoblotting
We homogenized the diaphragm samples using a Kontess Duall Homogenizer in mitochondrial isolation medium (in mM: sucrose 300, Tris 10, EGTA 1; pH 7.1), centrifuged the homogenate at 500 g for 7 min at 4°C (#5424 Eppendorf, Hauppauge, NY) and saved the supernatant for measurement of total protein content (DC Assay, Bio-Rad Laboratories, Hercules, CA) and SDS-PAGE and immunoblotting. We mixed the supernatant 1:3 with 4× Laemmli buffer (Bio-Rad Laboratories, Hercules, CA) with 0.35 M dithiothreitol, loaded approximately 15 µg of protein per lane into 4-20% Criterion™ TGX™ stain-free gels (Bio-Rad Laboratories, Hercules, CA), and ran electrophoresis for 50 min at 200 V with the apparatus surrounded by ice. We activated and scanned the gel in a Gel Doc EZ Imager (Bio-Rad Laboratories, Hercules, CA) and determined the total protein in each lane (Image Lab, Bio-Rad Laboratories, Hercules, CA). Subsequently, we transferred the proteins to a nitrocellulose membrane (GE Healthcare Life Sciences, Pittsburgh, PA) at a constant current (100 mA) overnight at 4°C. We blocked the membrane with Odyssey Blocking Buffer (LI-COR, Lincoln, NE) for 1 hr at room temperature. Then, we washed the membrane with Tris-buffered solution (TBS) for 5 min and incubated it with primary antibody for citrate synthase (ab96600; Abcam, Cambridge, MA) at 1:1,000 dilution and total OXPHOS antibody cocktail (ab110413; Abcam, Cambridge, MA) at 1:1,000 dilution for 1 hr at room temperature to detect citrate synthase and mitochondrial complexes I-V. Thereafter, we washed the membrane with TBS containing 0.1% of TWEEN®-20 (Sigma-Aldrich, St. Louis, MO) 4 × 5 min, followed by a TBS rinse for 5 min. We then incubated the membrane with secondary antibodies IRDye680 (926-68020; LI-COR, Lincoln, NE) at 1:40,000 dilution and IRDye800 (926-32211; LI-COR, Lincoln, NE) at 1:20,000 dilution for 1 hr in the dark, followed by a rinse as described above. We scanned the membrane in Odyssey CLx Infrared Imaging System (LI-COR, Lincoln, NE) and used Image Studio Lite Software (Li-COR, Lincoln, NE) to quantify the optical density of specific bands. We normalized the signal for each target protein to the total protein per lane, which were all within the linearity of the assays. We did not measure Nox4 protein abundance because there was no commercial antibody that reliably detected Nox4 – all antibodies tested showed a protein band in knockout animals, despite confirmed genotype and undetectable Nox4 mRNA.
For determination of myosin heavy chain and actin abundance and actin carbonylation, we homogenized the diaphragm samples in ice-cold high-salt protein isolation buffer (in mM: 300 NaCl, 100 NaH2PO4,
50 Na2HPO4, 10 Na4P2O7, 1 MgCl2, 10 EDTA, 1 dithiothreitol; pH 7.1) [37, 38] containing Halt protease inhibitor cocktail (Thermo Scientific, Waltham, MA). After homogenization, we centrifuged the samples at 10,000 g for 3 min at 4°C and used the supernatant for the assays. We measured total protein content with a Bradford assay (23246; Thermo Fisher Scientific, Waltham, MA), diluted the samples to similar protein contents, and mixed 1:1 with 2× Laemmli buffer (Bio-Rad Laboratories, Hercules, CA) containing 0.35 M dithiothreitol and heat-denatured for 4 min at 100°C. We loaded approximately 0.6 µg of total protein per lane, performed SDS-PAGE using Stain-FreeTM gel (Bio-Rad), activated and scanned the gel (Gel-Doc EZ System, Bio-Rad) for quantification of total protein (ImageLabTM, Bio-Rad), transferred to a nitrocellulose membrane (100 mA overnight), and used a commercial kit (OxiSelect STA-308; Cell Biolabs, San Diego, CA) to determine actin carbonylation [39]. Accordingly, we incubated the membrane in TBS containing 20% methanol for 5 min and washed in 2 N HCl for 5 min. We incubated the membrane with DNPH (2,4-dinitrophenylhydrazine) in 2 N HCl for derivatization of the carbonyl group for 5 min. Subsequently, we washed the membrane in 2 N HCl 3 × 5 min and TBS containing 50% methanol 5 × 5 min. We blocked the membrane with Odyssey Blocking Buffer (LI-COR, Lincoln, NE) for 1 hr, rinsed in TBS containing 0.1% of TWEEN®-20 (Sigma-Aldrich, St. Louis, MO) 4 × 5 min, and probed with primary antibody for DNP (2,4-dinitrophenol, 230801; Cell Biolabs, San Diego, CA) at 1:1,000 dilution. After washing steps, a secondary antibody IRDye800 (926-32211; LI-COR, Lincoln, NE) probed the membrane at 1:20,000 dilution for 1 hr in the dark, followed by washes and membrane scan in Odyssey CLx Infrared Imaging System (LI-COR, Lincoln, NE). Afterward, we exposed the same membrane to a primary antibody for actin (JLA20; Developmental Studies Hybridoma Bank, Iowa City, IA) at 1:1,000 dilution. After washing, we incubated the membrane with secondary antibody IRDye680 (926-68020; LI-COR, Lincoln, NE) at 1:40,000 dilution for 1 hr in the dark. We probed for actin and DNP on the same membrane using two fluorescence channels (λ = 700 and 800 nm for secondary antibodies). We used GelBandFitter [40] and Image Studio Lite Software (Li-COR, Lincoln, NE) to quantify the optical density of proteins and determined actin carbonylation as the carbonyl signal overlapping the actin band divided by total actin [39]. Actin carbonylation data are the average values of measurements in triplicate. We also quantified the abundance of myosin heavy chain based on the signal intensity of the band corresponding to 200-250 kDa of the Stain-Free gel.
Statistics
We used SigmaPlot v14.0 (Systat Software, San Jose, CA) to perform normality and equal variance tests and log-transformed non-parametric data before analysis with parametric tests. We used Prism v6
(GraphPad, La Jolla, CA) to run a two-way ANOVA with Bonferroni’s posthoc test (factor 1, strain: WT vs. Nox4 KO; factor 2, surgery: Sham vs. MI). In some cases, we performed posthoc tests when the two-way ANOVA interaction p-value was greater than 0.05 [41]. Because fiber cross-sectional area includes several measurements per animal, we applied a linear mixed model analysis to compare these variables among groups (SPSS v26, IBM, Armonk, NY). Where appropriate, we compared data with paired or unpaired Student’s t-test as needed. We used the Mann-Whitney rank-sum test for a failed normality test and Welch’s t-test in case of a failed equal variance testing. All statistics are from two-tailed tests, and we highlighted differences when p < 0.05. We opted to report exact p-values when feasible and follow recent recommendations for data interpretation [42, 43].
Results
We performed MI surgeries on 16 mice (WT n = 8, Nox4KO n = 8) and Sham surgeries on 10 mice (n = 5 per strain). Two WT mice died within 48 hrs of MI surgery. One Nox4KO mice assigned to the MI group had to be euthanized during surgery because of an accidental ventricular tear and excessive bleeding in the process of coronary artery ligation. All animals in the Sham group survived the duration of the study. Genotyping confirmed Nox4 knockout (Supplementary Fig. S1). There was no difference in infarct size between strains, and MI caused cardiac hypertrophy, evident by a ~30% increase in heart weight-to-tibial length (Table 1, Supplementary Fig. S2). MI caused a greater percent loss of body weight compared to Sham 72 hrs post-surgery (Table 1, Supplementary Fig. S3). Diaphragm Nox4 mRNA was elevated almost 80% in WT with MI but did not reach the threshold for statistical significance (in fold control: Sham = 1.00 ± 0.09, MI = 1.77 ± 0.65; p = 0.057). Nox4 mRNA was undetectable in Nox4KO mice.
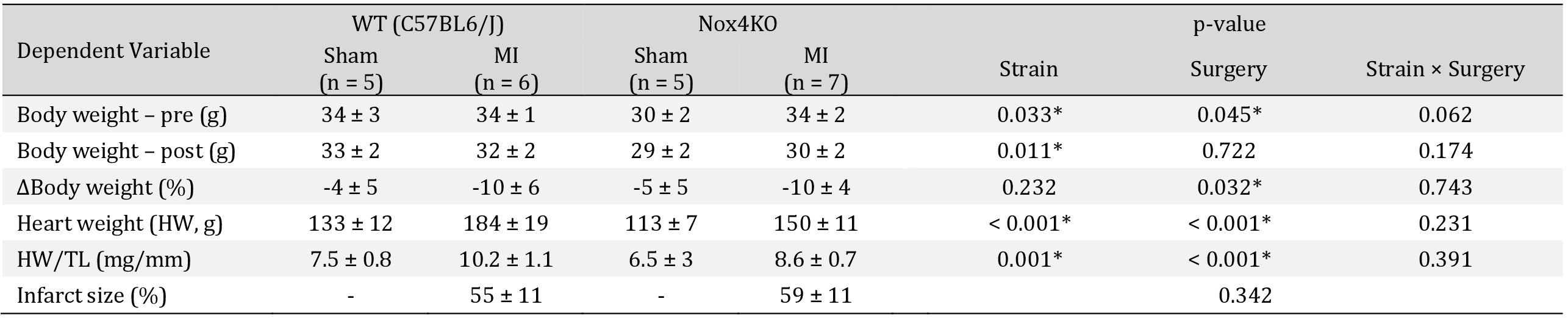
Animal characteristics. Each row contains p-values from two-way ANOVA for each factor and interaction for body weight, heart weight analysis. We performed unpaired Student's t-test for infarct size in MI WT vs. Nox4 KO. HW, heart weight; TL, tibial length. Data are means ± SD. *p < 0.05 by two-way ANOVA
Diaphragm atrophy, contractile dysfunction, and protein oxidation
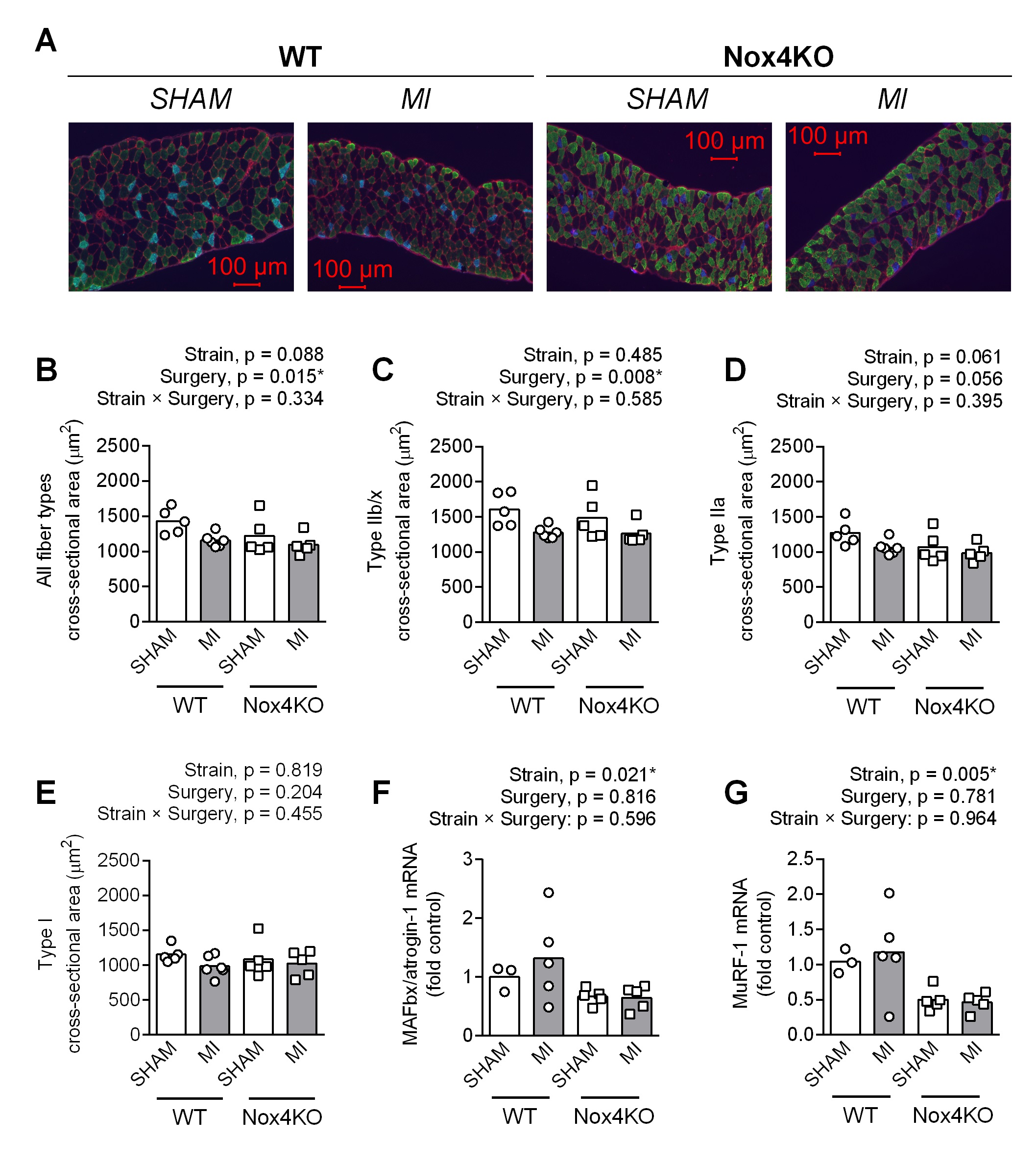
Diaphragm fiber cross-sectional area (CSA) was 15% lower in Nox4KO vs WT within Sham (Fig. 1B). MI decreased the overall fiber CSA by 10-20% in both WT and Nox4KO (Fig. 1B). Fiber type-specific analysis (Fig. 1C-E) showed that MI decreased type IIb/x fiber CSA by 20% in both WT and Nox4KO. Type IIa fiber CSA was lower post-MI than Sham, but the surgery effect on ANOVA did not reach the threshold for statistical significance (p = 0.056). There was no surgery or strain effect on type I CSA. Surprisingly, Nox4KO mice showed 30-50% lower mRNA abundance for MAFbx/atrogin-1 (Fig. 1F) and MuRF-1 (Fig. 1G). Analysis of fiber type percentage showed higher type IIa and lower type IIb/x fibers in Nox4KO vs. WT, whereas MI did not affect fiber type distribution (Supplementary Fig. S4).
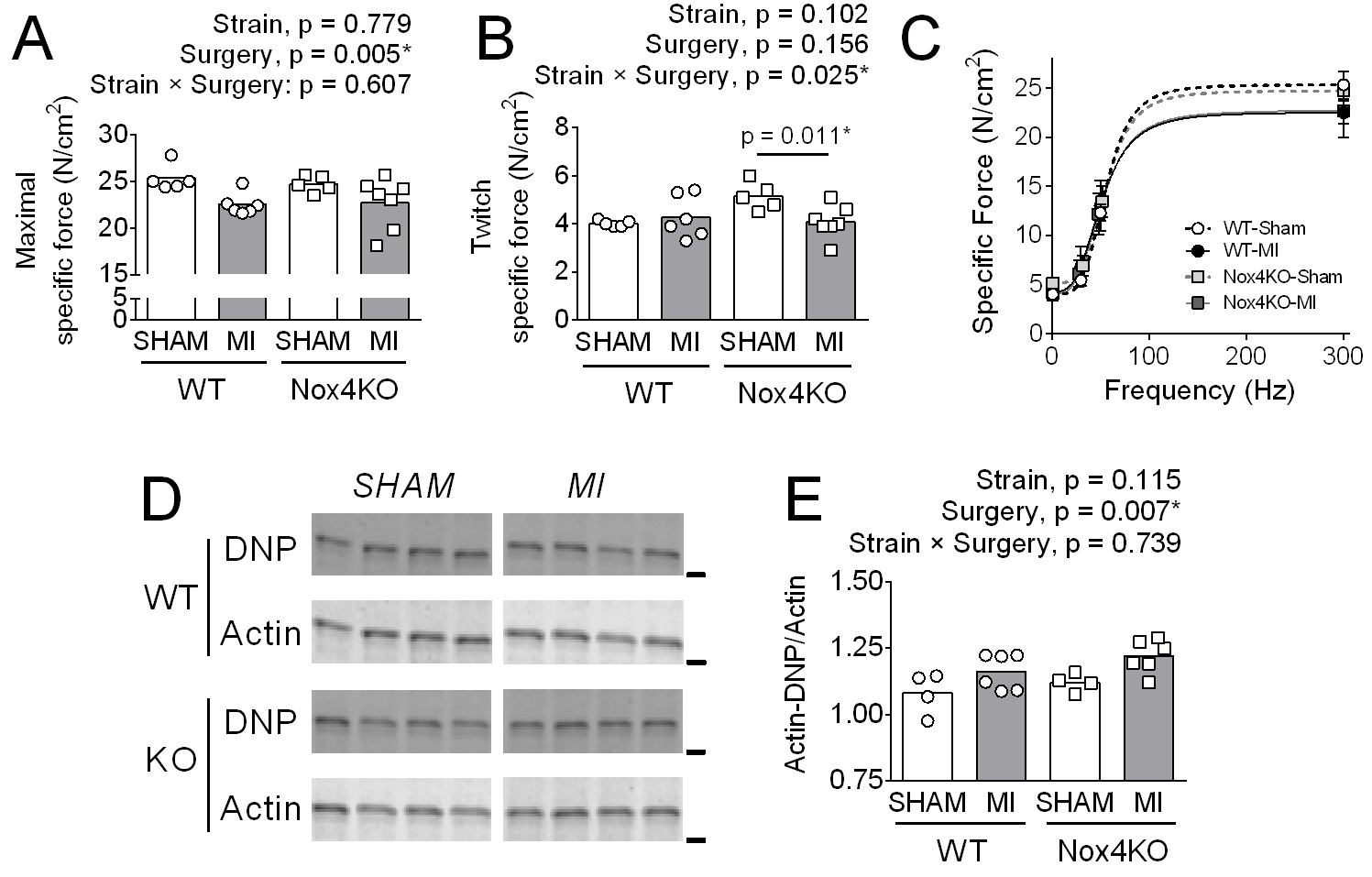
MI decreased maximal diaphragm specific force by 10% in both WT and Nox4KO (Fig. 2A). In Nox4KO, MI decreased twitch specific force by 20% (Fig. 2B). The decrease in force was accompanied by increased actin carbonylation in WT and Nox4KO MI vs. Sham
(Fig. 2C, D), and there were no changes in the most abundant thick (MyHC) and thin (actin) filament proteins with MI or Nox4 KO (Supplementary Fig. S5).
Mitochondrial proteins, respiration, and H2O2 emission
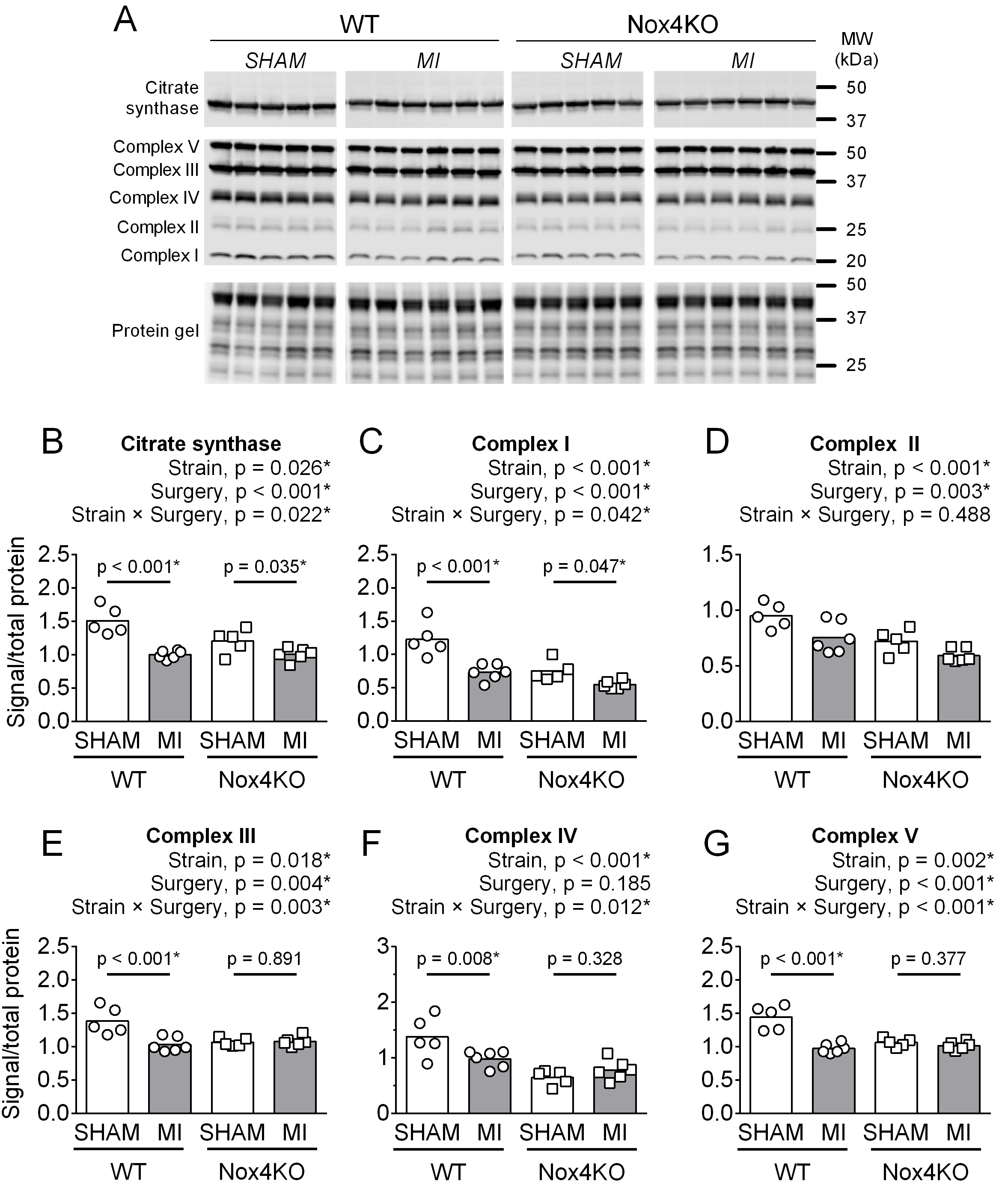
In WT mice, MI decreased the abundance of citrate synthase and protein subunits of the mitochondrial electron transport system (ETS) complex I-V compared to Sham (Fig. 3). Nox4KO decreased the abundance of citrate synthase and subunits of ETS complexes I-V compared to WT and did not prevent the MI-induced decrease in citrate synthase and subunits of ETS complexes I and II (Fig. 3B-D).
The effects of Nox4KO and MI on protein abundance of subunits of mitochondrial ETS complexes did not translate to lower mitochondrial O2 flux (Fig. 4A-D). MI had no detectable effect on diaphragm fiber JO2 (pmol/s/μg total protein), but Nox4KO displayed higher values than WT, particularly in Sham with complex I and I+II substrates during State 3 (ADP 500 µM). The JO2 normalized to citrate synthase abundance was higher post-MI (Fig. 4E-F), with differences determined mainly by WT Sham vs. MI. Nox4KO increased the respiratory control ratio (RCR) with 500 µM ADP (RCR; JO2 State 3-to-State 2), but there was no effect of MI on RCR (WT-Sham = 2.3 ± 0.4, WT-MI = 2.4 ± 0.3, Nox4KO-Sham = 3.0 ± 0.5, Nox4KO-MI = 3.3 ± 0.5; ANOVA strain p < 0.001, surgery p = 0.388, strain × surgery p = 0.753).
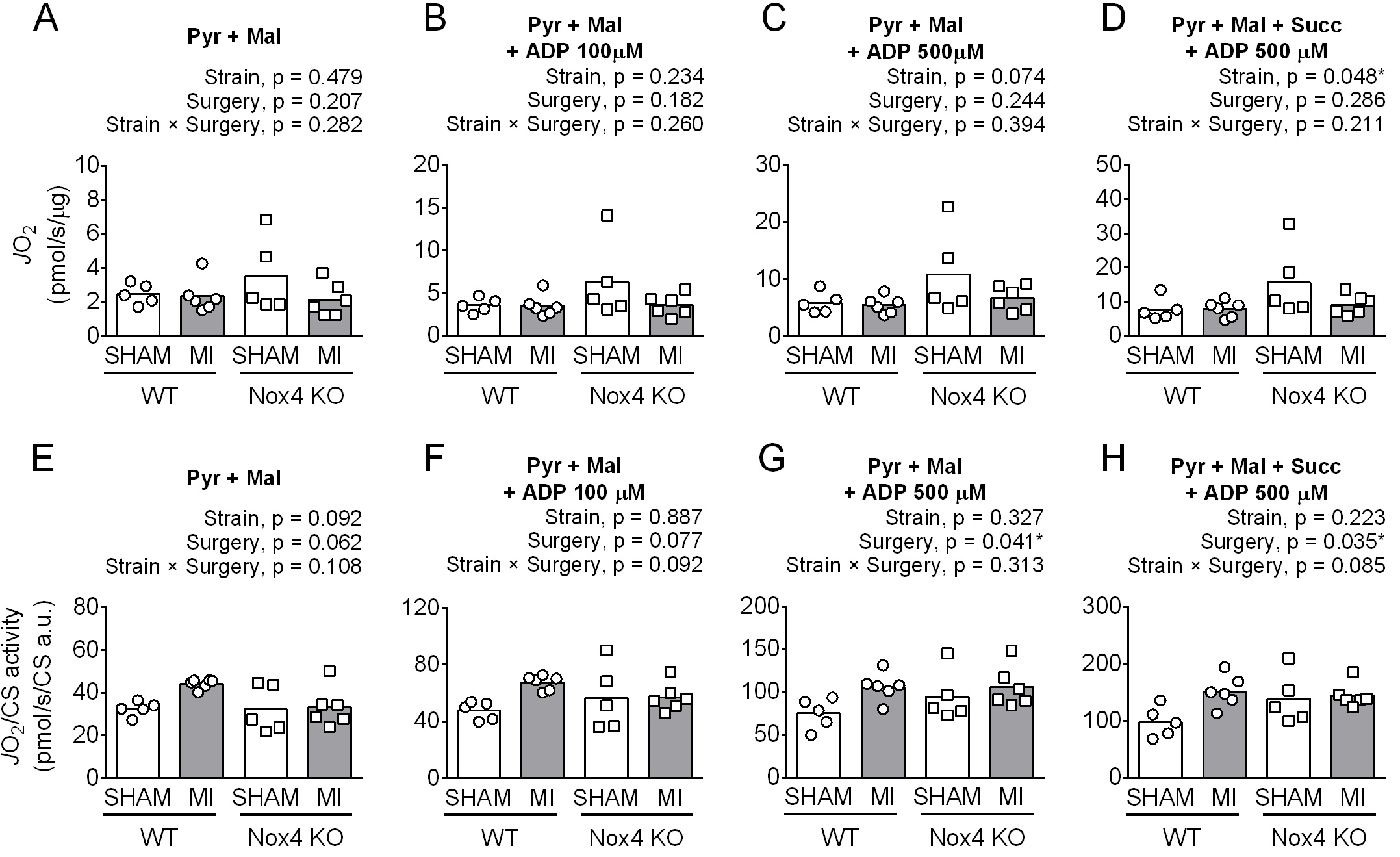
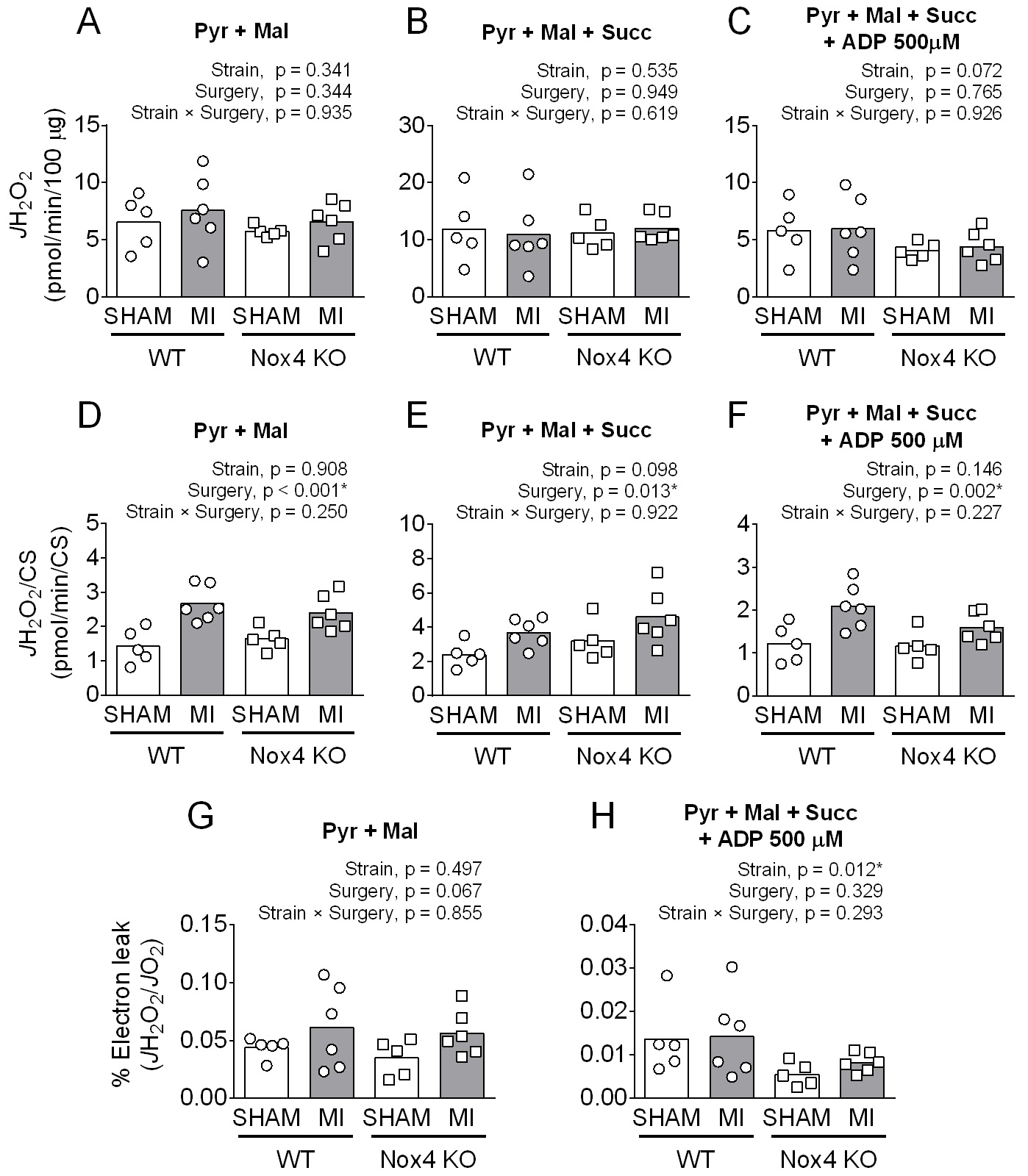
H2O2 emission
There were no differences in diaphragm JH2O2 (pmol/min/100 µg protein), except for complex I+II substrates in the presence of ADP with lower values in Nox4KO that did not reach statistical significance (Fig. 5A-C). However, JH2O2 normalized to citrate synthase abundance was 50-65% higher post-MI (Fig. 5D-F), independent of strains. Electron leak (JH2O2/JO2) in the absence of ADP was also higher post-MI for both strains but did not reach p < 0.05 (Fig. 5G). Nox4KO decreased the electron leak in the presence of ADP, but there were no differences between Sham and MI for both strains (Fig. 5H).
We would like to thank Deborah Morse for maintaining the colony of Nox4 knockout mice used in the study and Rachel Kelley for technical support.
Author Contributions
Designed the study and experiments: DH, TER, LFF; Performed experiments: DH, RAK, DRM, LFF; Data analysis and interpretation: DH, RAK, DRM, TER, KS, LFF; Provided essential resources: TER, KS, LFF; Drafted and edited the manuscript: DH, RAK, TER, KS, LFF; Approved the manuscript: DH, RAK, DRM, TER, KS, LFF.
Funding Sources
This study was funded by a University of Florida Research Foundation Professorship award, the American Physiological Society Giles Filley Award, University of Florida DRPD-ROF2020 grant, and NIH R01-HL130318 to L. Ferreira. T.E. Ryan was funded by NIH HL149704. AHA 20PRE35200047 funded R.A. Kumar.
Statement of Ethics
Our animal experiments conformed to internationally accepted standards and have been approved by the University of Florida Instistutional Animal Care and Use Committee protocol 201809075.
The authors declare they have no conflict of interests.
| 1 Ibanez B, Heusch G, Ovize M, Van de Werf F: Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 2015;65:1454-1471. https://doi.org/10.1016/j.jacc.2015.02.032 |
||||
| 2 Neves LM, Karsten M, Neves VR, Beltrame T, Borghi-Silva A, Catai AM: Relationship between inspiratory muscle capacity and peak exercise tolerance in patients post-myocardial infarction. Heart Lung 2012;41:137-145. https://doi.org/10.1016/j.hrtlng.2011.07.010 |
||||
| 3 Bowen TS, Mangner N, Werner S, Glaser S, Kullnick Y, Schrepper A, Doenst T, Oberbach A, Linke A, Steil L, Schuler G, Adams V: Diaphragm muscle weakness in mice is early-onset post-myocardial infarction and associated with elevated protein oxidation. J Appl Physiol (1985) 2015;118:11-19. https://doi.org/10.1152/japplphysiol.00756.2014 |
||||
| 4 Smith IJ, Roberts B, Beharry A, Godinez GL, Payan DG, Kinsella TM, Judge AR, Ferreira LF: Janus kinase inhibition prevents cancer- and myocardial infarction-mediated diaphragm muscle weakness in mice. Am J Physiol Regul Integr Comp Physiol 2016;310:R707-710. https://doi.org/10.1152/ajpregu.00550.2015 |
||||
| 5 Kelley RC, Ferreira LF: Diaphragm abnormalities in heart failure and aging: mechanisms and integration of cardiovascular and respiratory pathophysiology. Heart Fail Rev 2017;22:191-207. https://doi.org/10.1007/s10741-016-9549-4 |
||||
| 6 Woods PR, Olson TP, Frantz RP, Johnson BD: Causes of breathing inefficiency during exercise in heart failure. J Card Fail 2010;16:835-842. https://doi.org/10.1016/j.cardfail.2010.05.003 |
||||
| 7 Sieck GC, Fournier M: Diaphragm motor unit recruitment during ventilatory and nonventilatory behaviors. J Appl Physiol (1985) 1989;66:2539-2545. https://doi.org/10.1152/jappl.1989.66.6.2539 |
||||
| 8 Dempsey JA, Romer L, Rodman J, Miller J, Smith C: Consequences of exercise-induced respiratory muscle work. Respir Physiol Neurobiol 2006;151:242-250. https://doi.org/10.1016/j.resp.2005.12.015 |
||||
| 9 Du XJ, Cox HS, Dart AM, Esler MD: Sympathetic activation triggers ventricular arrhythmias in rat heart with chronic infarction and failure. Cardiovasc Res 1999;43:919-929. https://doi.org/10.1016/S0008-6363(99)00139-X |
||||
| 10 Liu Q, Chen D, Wang Y, Zhao X, Zheng Y: Cardiac autonomic nerve distribution and arrhythmia. Neural Regen Res 2012;7:2834-2841. | ||||
| 11 Del Rio R, Marcus NJ, Schultz HD: Carotid chemoreceptor ablation improves survival in heart failure: rescuing autonomic control of cardiorespiratory function. J Am Coll Cardiol 2013;62:2422-2430. https://doi.org/10.1016/j.jacc.2013.07.079 |
||||
| 12 Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM: Inflammation in myocardial diseases. Circ Res 2012;110:126-144. https://doi.org/10.1161/CIRCRESAHA.111.243170 |
||||
| 13 Leenen FH, Skarda V, Yuan B, White R: Changes in cardiac ANG II postmyocardial infarction in rats: effects of nephrectomy and ACE inhibitors. Am J Physiol 1999;276:H317-325. https://doi.org/10.1152/ajpheart.1999.276.1.H317 |
||||
| 14 Kadoguchi T, Shimada K, Koide H, Miyazaki T, Shiozawa T, Takahashi S, Aikawa T, Ouchi S, Kitamura K, Sugita Y, Hamad AS, Kunimoto M, Sato-Okabayashi Y, Akita K, Isoda K, Daida H: Possible Role of NADPH Oxidase 4 in Angiotensin II-Induced Muscle Wasting in Mice. Front Physiol 2018;9:340. https://doi.org/10.3389/fphys.2018.00340 |
||||
| 15 van Hees HW, van der Heijden HF, Ottenheijm CA, Heunks LM, Pigmans CJ, Verheugt FW, Brouwer RM, Dekhuijzen PN: Diaphragm single-fiber weakness and loss of myosin in congestive heart failure rats. Am J Physiol Heart Circ Physiol 2007;293:H819-828. https://doi.org/10.1152/ajpheart.00085.2007 |
||||
| 16 Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Lusini L, Milzani A, Di Simplicio P, Colombo R: Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic Biol Med 2001;31:1075-1083. https://doi.org/10.1016/S0891-5849(01)00690-6 |
||||
| 17 Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK: Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res 2018;122:877-902. https://doi.org/10.1161/CIRCRESAHA.117.311401 |
||||
| 18 Manea A, Tanase LI, Raicu M, Simionescu M: Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun 2010;396:901-907. https://doi.org/10.1016/j.bbrc.2010.05.019 |
||||
| 19 Xia F, Wang C, Jin Y, Liu Q, Meng Q, Liu K, Sun H: Luteolin protects HUVECs from TNF-alpha-induced oxidative stress and inflammation via its effects on the Nox4/ROS-NF-kappaB and MAPK pathways. J Atheroscler Thromb 2014;21:768-783. https://doi.org/10.5551/jat.23697 |
||||
| 20 Sun QA, Hess DT, Nogueira L, Yong S, Bowles DE, Eu J, Laurita KR, Meissner G, Stamler JS: Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor-Ca2+ release channel by NADPH oxidase 4. Proc Natl Acad Sci U S A 2011;108:16098-16103. https://doi.org/10.1073/pnas.1109546108 |
||||
| 21 Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, Jackson MJ: Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 2013;18:603-621. https://doi.org/10.1089/ars.2012.4623 |
||||
| 22 Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J: Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 2010;106:1253-1264. https://doi.org/10.1161/CIRCRESAHA.109.213116 |
||||
| 23 Dasgupta A, Shukla SK, Vernucci E, King RJ, Abrego J, Mulder SE, Mullen NJ, Graves G, Buettner K, Thakur R, Murthy D, Attri KS, Wang D, Chaika NV, Pacheco CG, Rai I, Engle DD, Grandgenett PM, Punsoni M, Reames BN, et al.: SIRT1-NOX4 signaling axis regulates cancer cachexia. J Exp Med 2020;217:e20190745. https://doi.org/10.1084/jem.20190745 |
||||
| 24 Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, Trivedi T, Charkhzarrin S, Khatiwada P, Wronska A, Haynes A, Benassi MS, Witzmann FA, Zhen G, Wang X, Cao X, et al.: Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med 2015;21:1262-1271. https://doi.org/10.1038/nm.3961 |
||||
| 25 Mongue-Din H, Patel AS, Looi YH, Grieve DJ, Anilkumar N, Sirker A, Dong X, Brewer AC, Zhang M, Smith A, Shah AM: NADPH Oxidase-4 Driven Cardiac Macrophage Polarization Protects Against Myocardial Infarction-Induced Remodeling. JACC Basic Transl Sci 2017;2:688-698. https://doi.org/10.1016/j.jacbts.2017.06.006 |
||||
| 26 Stevenson MD, Canugovi C, Vendrov AE, Hayami T, Bowles DE, Krause KH, Madamanchi NR, Runge MS: NADPH Oxidase 4 Regulates Inflammation in Ischemic Heart Failure: Role of Soluble Epoxide Hydrolase. Antioxid Redox Signal 2019;31:39-58. https://doi.org/10.1089/ars.2018.7548 |
||||
| 27 Hood DA, Memme JM, Oliveira AN, Triolo M: Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu Rev Physiol 2019;81:19-41. https://doi.org/10.1146/annurev-physiol-020518-114310 |
||||
| 28 Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM: NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A 2010;107:18121-18126. https://doi.org/10.1073/pnas.1009700107 |
||||
| 29 Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP: Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 2012;110:1217-1225. https://doi.org/10.1161/CIRCRESAHA.112.267054 |
||||
| 30 Emde B, Heinen A, Godecke A, Bottermann K: Wheat germ agglutinin staining as a suitable method for detection and quantification of fibrosis in cardiac tissue after myocardial infarction. Eur J Histochem 2014;58:2448. https://doi.org/10.4081/ejh.2014.2448 |
||||
| 31 Wen Y, Murach KA, Vechetti IJ, Jr., Fry CS, Vickery C, Peterson CA, McCarthy JJ, Campbell KS: MyoVision: software for automated high-content analysis of skeletal muscle immunohistochemistry. J Appl Physiol (1985) 2018;124:40-51. https://doi.org/10.1152/japplphysiol.00762.2017 |
||||
| 32 Close RI: Dynamic properties of mammalian skeletal muscles. Physiol Rev 1972;52:129-197. https://doi.org/10.1152/physrev.1972.52.1.129 |
||||
| 33 Hahn D, Kumar RA, Ryan TE, Ferreira LF: Mitochondrial Respiration and H2O2 Emission in Saponin-permeabilized Murine Diaphragm Fibers: Optimization of Fiber Separation and Comparison to Limb Muscle. Am J Physiol Cell Physiol 2019:317:C665-C673. https://doi.org/10.1152/ajpcell.00184.2019 |
||||
| 34 Sieck GC, Ferreira LF, Reid MB, Mantilla CB: Mechanical properties of respiratory muscles. Compr Physiol 2013;3:1553-1567. https://doi.org/10.1002/cphy.c130003 |
||||
| 35 Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, Neufer PD: Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J 2011;437:215-222. https://doi.org/10.1042/BJ20110366 |
||||
| 36 Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F, Hey-Mogensen M: Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 2012;590:3349-3360. https://doi.org/10.1113/jphysiol.2012.230185 |
||||
| 37 Cosper PF, Leinwand LA: Myosin Heavy Chain is not Selectively Decreased in Murine Cancer Cachexia. Int J Cancer 2012;130:2722-2727. https://doi.org/10.1002/ijc.26298 |
||||
| 38 Roberts BM, Frye GS, Ahn B, Ferreira LF, Judge AR: Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem Biophys Res Commun 2013;435:488-492. https://doi.org/10.1016/j.bbrc.2013.05.018 |
||||
| 39 Coblentz PD, Ahn B, Hayward LF, Yoo JK, Christou DD, Ferreira LF: Small-hairpin RNA and pharmacological targeting of neutral sphingomyelinase prevent diaphragm weakness in rats with heart failure and reduced ejection fraction. Am J Physiol Lung Cell Mol Physiol 2019;316:L679-L690. https://doi.org/10.1152/ajplung.00516.2018 |
||||
| 40 Mitov MI, Greaser ML, Campbell KS: GelBandFitter--a computer program for analysis of closely spaced electrophoretic and immunoblotted bands. Electrophoresis 2009;30:848-851. https://doi.org/10.1002/elps.200800583 |
||||
| 41 Wei J, Carroll RJ, Harden KK, Wu G: Comparisons of treatment means when factors do not interact in two-factorial studies. Amino Acids 2012;42:2031-2035. https://doi.org/10.1007/s00726-011-0924-0 |
||||
| 42 Amrhein V, Greenland S, McShane B: Scientists rise up against statistical significance. Nature 2019;567:305-307. https://doi.org/10.1038/d41586-019-00857-9 |
||||
| 43 Curran-Everett D: Evolution in statistics: P values, statistical significance, kayaks, and walking trees. Adv Physiol Educ 2020;44:221-224. https://doi.org/10.1152/advan.00054.2020 |
||||
| 44 Sutton MG, Sharpe N: Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 2000;101:2981-2988. https://doi.org/10.1161/01.CIR.101.25.2981 |
||||
| 45 Rubin SA, Fishbein MC, Swan HJ: Compensatory hypertrophy in the heart after myocardial infarction in the rat. J Am Coll Cardiol 1983;1:1435-1441. https://doi.org/10.1016/S0735-1097(83)80046-1 |
||||
| 46 Murdoch CE, Zhang M, Cave AC, Shah AM: NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res 2006;71:208-215. https://doi.org/10.1016/j.cardiores.2006.03.016 |
||||
| 47 Matsushima S, Kuroda J, Ago T, Zhai P, Ikeda Y, Oka S, Fong GH, Tian R, Sadoshima J: Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ Res 2013;112:1135-1149. https://doi.org/10.1161/CIRCRESAHA.111.300171 |
||||
| 48 Zhu E, Sassoon CS, Nelson R, Pham HT, Zhu L, Baker MJ, Caiozzo VJ: Early effects of mechanical ventilation on isotonic contractile properties and MAF-box gene expression in the diaphragm. J Appl Physiol (1985) 2005;99:747-756. https://doi.org/10.1152/japplphysiol.00126.2005 |
||||
| 49 Adams V, Bowen TS, Werner S, Barthel P, Amberger C, Konzer A, Graumann J, Sehr P, Lewis J, Provaznik J, Benes V, Buttner P, Gasch A, Mangner N, Witt CC, Labeit D, Linke A, Labeit S: Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J Cachexia Sarcopenia Muscle 2019;10:1102-1115. https://doi.org/10.1002/jcsm.12448 |
||||
| 50 Ottenheijm CA, Heunks LM, Li YP, Jin B, Minnaard R, van Hees HW, Dekhuijzen PN: Activation of the ubiquitin-proteasome pathway in the diaphragm in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006;174:997-1002. https://doi.org/10.1164/rccm.200605-721OC |
||||
| 51 Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW: Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994;74:1141-1148. https://doi.org/10.1161/01.RES.74.6.1141 |
||||
| 52 von Lohneysen K, Noack D, Hayes P, Friedman JS, Knaus UG: Constitutive NADPH oxidase 4 activity resides in the composition of the B-loop and the penultimate C terminus. J Biol Chem 2012;287:8737-8745. https://doi.org/10.1074/jbc.M111.332494 |
||||
| 53 Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S: Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med 2013;19:101-106. https://doi.org/10.1038/nm.3019 |
||||
| 54 Youm TH, Woo SH, Kwon ES, Park SS: NADPH Oxidase 4 Contributes to Myoblast Fusion and Skeletal Muscle Regeneration. Oxid Med Cell Longev 2019;2019:3585390. https://doi.org/10.1155/2019/3585390 |
||||
| 55 Plecita-Hlavata L, Jaburek M, Holendova B, Tauber J, Pavluch V, Berkova Z, Cahova M, Schroder K, Brandes RP, Siemen D, Jezek P: Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes 2020;69:1341-1354. https://doi.org/10.2337/db19-1130 |
||||
| 56 TeSlaa T, Bartman CR, Jankowski CSR, Zhang Z, Xu X, Xing X, Wang L, Lu W, Hui S, Rabinowitz JD: The Source of Glycolytic Intermediates in Mammalian Tissues. Cell Metab 2021;33:367-378.e5. https://doi.org/10.1016/j.cmet.2020.12.020 |
||||
| 57 Bagni MA, Colombini B, Nocella M, Pregno C, A SC, Rassier DE: The effects of fatigue and oxidation on contractile function of intact muscle fibers and myofibrils isolated from the mouse diaphragm. Sci Rep 2019;9:4422. https://doi.org/10.1038/s41598-019-39353-5 |
||||
| 58 Ferreira LF, Laitano O: Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med 2016;98:18-28. https://doi.org/10.1016/j.freeradbiomed.2016.05.011 |
||||
| 59 Reid MB, Li YP: Tumor necrosis factor-alpha and muscle wasting: a cellular perspective. Respir Res 2001;2:269-272. https://doi.org/10.1186/rr67 |
||||
| 60 Dikalov SI, Nazarewicz RR: Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 2013;19:1085-1094. https://doi.org/10.1089/ars.2012.4604 |
||||
| 61 Barthel P, Wensel R, Bauer A, Muller A, Wolf P, Ulm K, Huster KM, Francis DP, Malik M, Schmidt G: Respiratory rate predicts outcome after acute myocardial infarction: a prospective cohort study. Eur Heart J 2013;34:1644-1650. https://doi.org/10.1093/eurheartj/ehs420 |
||||
| 62 Bernard K, Logsdon NJ, Miguel V, Benavides GA, Zhang J, Carter AB, Darley-Usmar VM, Thannickal VJ: NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J Biol Chem 2017;292:3029-3038. https://doi.org/10.1074/jbc.M116.752261 |
||||
| 63 Koziel R, Pircher H, Kratochwil M, Lener B, Hermann M, Dencher NA, Jansen-Durr P: Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem J 2013;452:231-239. https://doi.org/10.1042/BJ20121778 |
||||
| 64 Zuo L, Best TM, Roberts WJ, Diaz PT, Wagner PD: Characterization of reactive oxygen species in diaphragm. Acta Physiol (Oxf) 2015;213:700-710. https://doi.org/10.1111/apha.12410 |
||||
| 65 McLaughlin KL, Hagen JT, Coalson HS, Nelson MAM, Kew KA, Wooten AR, Fisher-Wellman KH: Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci Rep 2020;10:17599. https://doi.org/10.1038/s41598-020-74718-1 |
||||