×
![]()
Corresponding Author: Xiaomin Bao
Department of Molecular Biosciences, Northwestern University,
Hogan 2100, 2205 Tech Drive, Evanston, IL 60208, USA
Tel. +1-847-467-0418, E-Mail xiaomin.bao@northwestern.edu
Oh, the Mutations You’ll Acquire!
A Systematic Overview of Cutaneous Squamous Cell Carcinoma
Stephenie Drolla Xiaomin Baoa,b,c
aDepartment of Molecular Biosciences, Northwestern University, Evanston, IL, USA, bDepartment of Dermatology, Northwestern University, Chicago, IL, USA, cRobert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL, USA
Introduction
An invasive type of skin cancer, cSCC is the second-most common cancer worldwide. cSCC represents an incredible emotional, physical, and financial burden for patients and the healthcare system. In the USA alone, cSCC hospitalizes 6.2 per 100,000 people with an average stay of 5.8 days and cost of $66,000 [1] and causes an estimated 15,000 annual deaths [2]. This represents twice the melanoma death rate and matches deaths due to brain, esophageal, oral, or ovarian cancers [3]. Alarmingly, the incidence of cSCC continues to rise [4]. While new therapies are being developed to treat advanced cSCC, this life-threatening condition remains challenging to cure. cSCC is a multifaceted disease with numerous factors influencing its initiation and progression. Rather than focusing on a particular gene or pathway, this review will assess the layers of complexity. Cumulatively, this review will discuss the characteristics of cSCC, dysregulation of signaling pathways, alterations to critical nuclear effectors, and the resulting disorganization of the extracellular environment (Fig. 1). Where possible, we will highlight interactions between these modules, approved drugs, and novel therapeutic targets.
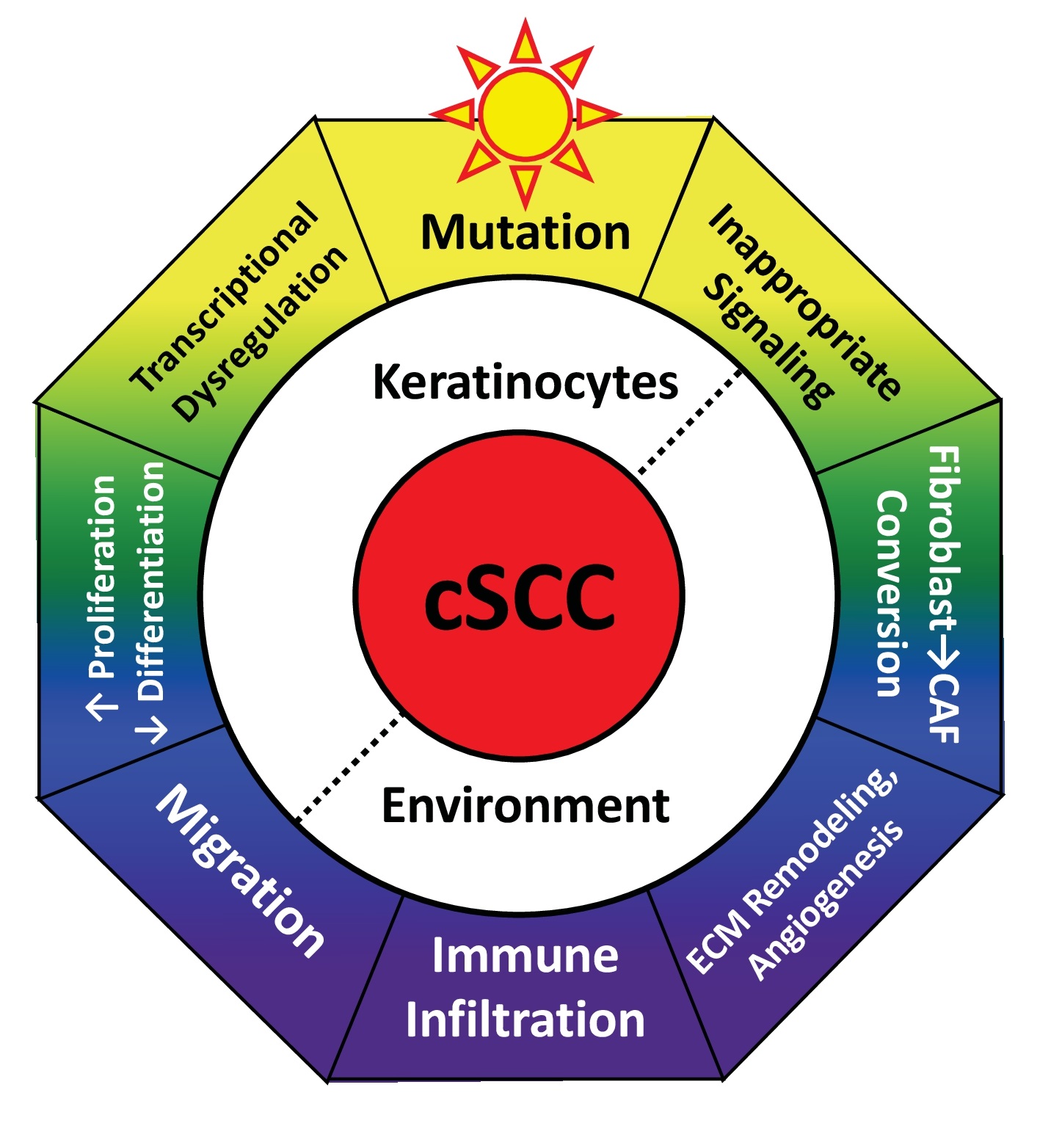
Characteristics of cSCC
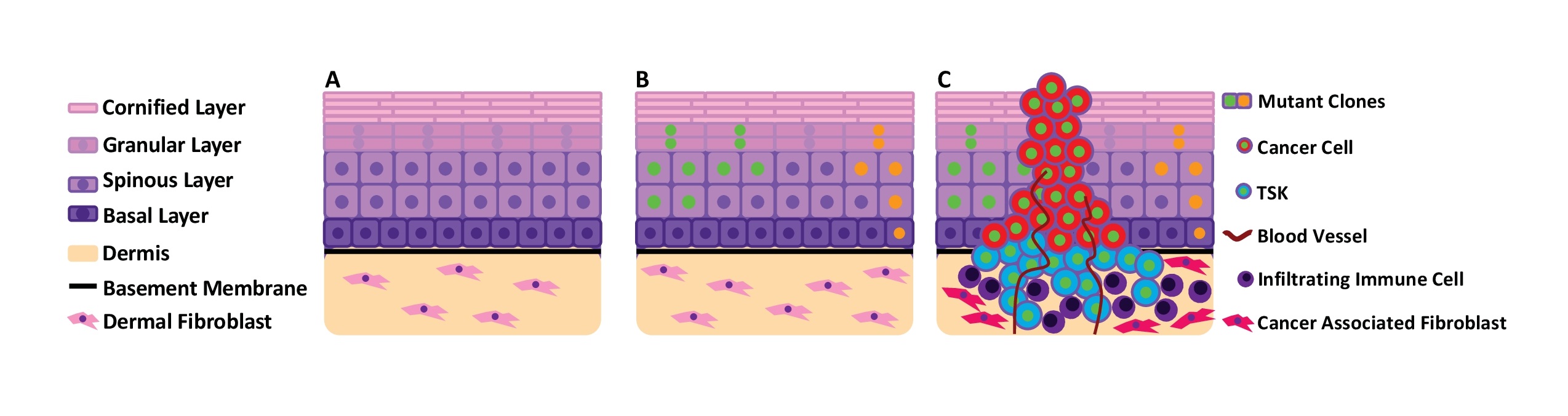
cSCC develops from skin epidermis, a critical physical and immune barrier protecting the human body. Epidermis is composed primarily of keratinocytes organized into multiple layers (termed stratified squamous epithelium). In homeostatic epidermis, keratinocyte proliferation is restricted to the basal layer, which is anchored to the basement membrane, a mechanical barrier of extracellular proteins which separate the epidermal and dermal components of skin. During differentiation, keratinocytes migrate to the upper layers (spinous, granular, and cornified strata), withdraw from the cell cycle, and sequentially activate genes allowing the production of proteins and lipids that contribute to barrier function (Fig. 2a) [5]. Dysregulation of keratinocyte proliferation and differentiation is fundamental to cSCC initiation and progression.
cSCC features the highest mutational burden of all cancers, with a median of 45.2 mutations per megabase (Mb) of genomic DNA [6, 7]. These mutations are substantially enriched in C > T or CC > TT substitutions, the characteristic ultraviolet (UV) mutation signature. Thus, UV radiation is considered the chief epidermal carcinogen [2]. Notably, these mutations occur in morphologically normal skin, but at a lower frequency of 2-6 coding mutations per Mb, where they drive dynamic, temporal, and spatial genetic mosaicism (Fig. 2b) [8]. Clones containing cancer-driver mutations (such as TP53, NOTCH, and RAS) undergo strong positive selection and expand in size [9, 10]. Remarkably, the mutational burden accumulated in normal epidermis is greater than that of many solid tumors, yet epidermis forestalls malignant transformation for decades [10]. How epidermis can tolerate such a high level of mutation remains unclear. Yet, this high mutational burden complicates identification of bona fide cSCC drivers and the development of targeted therapeutics, despite 60% of cSCC tumors potentially harboring mutations targetable by an existing small molecule therapy developed for other cancers [11].
The architecture of cSCC tumors is highly complex, composed of keratinocytes and a variety of stromal cells. At the invasive front of cSCC, a population of CD133+/CD45- keratinocytes (~1% of cells) act as tumor initiating cells (cancer stem cells). Xenograft of these sorted keratinocytes reliably replicates the histology of the original tumor [12]. Additionally, recent single-cell RNA sequencing of cSCC tumors also identified a unique subpopulation of tumor-specific keratinocytes (TSKs) at the invasive front (Fig. 2c). TSKs demonstrate strong upregulation of genes related to cell motility and extracellular matrix remodeling. Spatial gene-expression analysis identified proximity of TSKs with stromal cells (cancer-associated fibroblasts and vascular cells) as crucial for cancer progression [13]. Thus, TSKs can function as signaling hubs among different cell types in cSCC. In addition to TSKs at the invasive front, cSCC contains subpopulations of keratinocytes similar to normal tissue, but these cells generally exhibit altered metabolic and immune response, decreased apoptosis and differentiation, and epithelial-to-mesenchymal transition (EMT) [13]. Interestingly, cSCC cell lines also possess TSKs after being xenografted to mice, suggesting that TSKs may not necessarily have additional driver mutations compared to the other populations. Rather, the unique gene expression patterns of TSKs are likely to be induced and enhanced by the local environment.
The majority (65-80%) of cSCC develops from premalignant lesions called actinic keratosis (AK) and Bowen’s disease (BD). AK presents as a small, red, scaly patch, and forms when dysplastic keratinocytes proliferate. AK represents the third most common reason for dermatological consultation since the lesions are cosmetically displeasing and sometimes painful. BD tends to be redder, scalier, and slow-growing lesions characterized by full-thickness dysplasia that does not breach the basement membrane [14], sometimes referred to as “cSCC in situ.” While estimates vary, a single AK poses a 10% or 20% risk progressing to cSCC in immunocompetent and immunocompromised patients respectively while a BD lesion poses a 16.3% risk [14-16]. Treatments for AK and BD include: topical drugs (5-fluorouracil, imiquimod, ingenol mebutate, diclofenac), photodynamic therapy, and removal (surgical, cryotherapy, laser) [16]. While treatment reduces lesion size and number, patients are burdened by significant skin irritation, length of treatment, and lesion recurrence. For prophylaxis, oral retinoids likely provide no benefit, but proper sunscreen use reduces AK formation [16]. Oral nicotinamide promotes DNA repair by preventing ATP depletion after UV exposure. This safe and cheap drug reduces AK lesions [17] and cSCC occurrence by 30% in immunocompetent patients [18]. AK and BD represent a precancerous condition, but lesion growth and bleeding may suggest progression to cSCC.
Compared to the immunocompetent population, transplant patients have a dramatically increased (32-198 fold) risk of developing aggressive, relapsing cSCC [19]. While immunosuppression prevents graft rejection, it increases cSCC risk, which then threatens graft viability. In addition to reducing immune surveillance, calcineurin inhibitors and azathioprine, directly contribute to increased mutational burden. Azathioprine photosensitizes skin and increases reactive oxygen species (ROS) production upon UV exposure [20]. Calcineurin inhibitors increase cSCC occurrence by inhibiting the nucleotide and base excision DNA repair pathways [21]. Switching from calcineurin to mTOR inhibition halved skin cancer rates in transplant patients [22], as mTOR inhibitors can also suppress DNA damage, proliferation and angiogenesis [23]. Prevention and treatment of cSCC in immunocompromised patients represents a critical and growing clinical problem. Prophylactic retinoids (vitamin A derivatives) [24] and capecitabine (inhibitor of DNA repair) are currently prescribed off-label for AK and cSCC in transplant patients. Meta-analyses suggest some efficacy but also serious adverse reactions. No controlled clinical trial has evaluated prophylaxis efficacy and safety [25].
Until recently, treatment for advanced cSCC was limited to surgery and off-label therapies that lacked robust evidence of efficacy or safety. Surgical removal is still the frontline treatment for cSCC, but results in a 3-8% recurrence rate, even when the original tumor is properly excised. As cSCC frequently occurs on the face, surgery can be disfiguring. However, tumor placement, advancement, and patient health can prohibit surgical intervention [26]. Therefore, adjuvant radiotherapy is recommended for immunosuppression, incomplete excision, metastasis, perineural invasion, and recurrence [24, 27]. Local recurrence, lymph node metastasis, and distant metastases occur in 3-14%, 3.7-16%, and 5% of patients respectively [28-30]. Risk factors for recurrence and metastasis include: large tumor size, perineural or subcutaneous fat invasion, poor differentiation, and location on the temple, ear, or lip [31].
Alterations to Signaling Pathways
Cell growth, motility, and survival are strictly controlled by environmental cues in homeostatic tissue. These cues are received by cell surface receptors and processed by different signaling pathways to trigger specific cellular responses. A number of mutations have been identified in distinct components of pathways, bypassing signaling-regulated restrictions to promote uncontrolled growth. To illustrate the complexity of alterations in cSCC, this section will primarily focus on the epidermal growth factor receptor (EGFR) signaling pathway, but will also discuss the roles of NOTCH and TGFβ.
EGFR Pathway
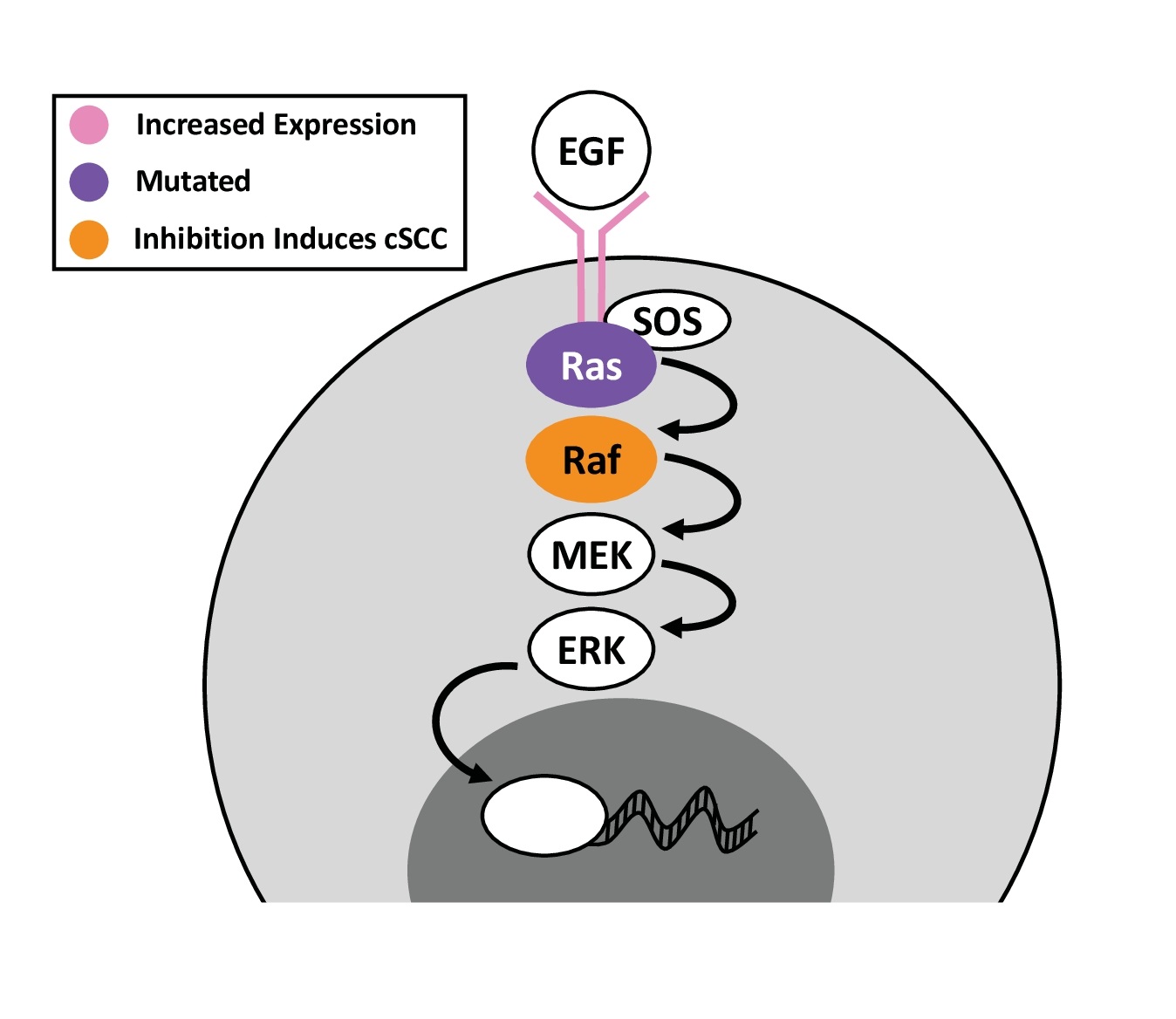
EGFR. The EGFR pathway plays crucial roles in regulating cell growth and survival. Four genes encode EGFR receptors in the human genome, but only three are expressed by normal keratinocytes: EGFR (ERBB1), HER2 (ERBB2), and HER3 (ERBB3). Seven ligands for EGFR have been identified with varying receptor affinity [32]. Upon activation, EGFR dimerizes and functions as tyrosine kinase to trigger downstream kinase cascades (Fig. 3). Interestingly, UV radiation activates EGFR by preventing its inhibitory dephosphorylation, leading to cyclin D activation and p53 suppression, which induce epidermal hyperplasia. UV radiation can also induce nuclear translocation of EGFR. UV-induced EGFR activation is a unique feature of epidermal tissue [33]. EGFR inhibition reduces UV-induced erythema, hyperplasia, lymphocyte infiltration, cytokine production, and tumor size [34-37].
EGFR copy-number gains occur frequently in cSCC, but also in AK [38], suggesting that the pathway is altered early during cSCC carcinogenesis. Immunohistochemistry (IHC) of cSCC consistently reveals EGFR overexpression (Fig. 3), and aberrant cytoplasmic staining [38-41]. Overexpression independently predicts disease progression and is more prevalent in metastatic than primary tumors [40, 42]. Moreover, a recurrent cSCC-specific EGFR-PPARGC1A fusion was recently identified in 30% of the studied tumors. The fusion protein is constitutively phosphorylated and dimerizes with wild-type EGFR, inducing activation. Cells expressing the EGFR-PPARGC1A fusion form even larger tumors than cells overexpressing EGFR while PPARGC1A overexpression does not induce tumors [43]. In contrast, EGFR mutation is relatively rare, affecting about 4.9% of cSCC cases [44].
Given EGFR’s frequent alteration in cSCC and its critical roles in cancer progression, EGFR represents an attractive therapeutic target with both antibodies and small molecule inhibitors being developed. Cetuximab, a monoclonal antibody targeting EGFR, was approved in 2006 for advanced cSCC of the head and neck. Used alone or with other therapy for unresectable and metastatic cSCC, cetuximab produces an overall response rate around 50% with an acceptable safety profile, even in elderly patients [45-47]. Another EGFR antibody, panitumumab, in combination with the talimogene laherparepvec vaccine (a virus-based melanoma therapy) is currently in phase I trial. On the small-molecule inhibitor front, recent phase II trials of lapatinib and gefitinib produced results only in a subset of patients. Two weeks of lapatinib reduced tumor volume in only 2 of 8 patients. However, two months after treatment, 7 of 8 patients did experience remission of concurrent AK [48]. Gefitinib induced partial response in 6 of 37 and stable disease in 13 of 37 patients with a median duration of 31.4 months [49]. The small-molecule-inhibitor, erlotinib failed to achieve an acceptable response rate in phase II trials [50]. Recently, preclinical screening identified DUBs-IN-3 as a potential cSCC drug. DUBs-IN-3 inhibits USP8, a component of the ubiquitin pathway that shields EGFR and other growth factor receptors from degradation. DUBs-IN-3 treatment reduces EGFR protein and preferentially kills cSCC cells. As DUBs-IN-3 multiple receptors, it may aid in overcoming drug resistance [51].
RAS. EGFR activation leads to phosphorylation and binding of Ras adaptor proteins, docking of guanine nucleotide exchange factors (GEFs), and activation of the Ras GTPase. Activated Ras serves as a signaling hub to activate several downstream kinases. There are three paralogs: H-Ras, K-Ras, and N-Ras. These paralogs are required, but redundant during epidermal development. H-Ras null mice are indistinguishable from wild-type littermates, as K-Ras and N-Ras can compensate for H-Ras in developmental processes [52]. Triple knockout of all three Ras paralogs in epidermal tissue inhibits both proliferation and differentiation, resulting in post-natal lethality. These triple-knockout keratinocytes exhibit G1-arrested senescence, downregulate p63 and c-MYC, and express early, but not late differentiation markers. Constitutive ERK2 activation can rescue proliferation of Ras-null cells and restores c-Myc and p63 expression [53]. These findings highlight the central role of Ras and its downstream signaling in controlling keratinocyte proliferation and differentiation.
Ras mutations affect a small subset of cSCC, with frequencies of: 6-16% H-Ras, 13% K-Ras, and 5% N-Ras (Fig. 3) [7, 44, 54, 55]. Much of the research exploring the role of Ras in cSCC has focused on H-Ras. Expression of H-RasG12V in keratinocytes causes oncogene-induced senescence [56]. However, in combination with additional oncogenic agents or mutations, Ras can potently drive rapid and aggressive cSCC progression. In the classic DMBA (7,12-dimethylbenz [a]anthracene) plus TPA (12-O-tetradecanoylphorbol-13-acetate) mouse model of cSCC, the mutagen DMBA promotes H-Ras mutation in 95% of tumors while repeated application TPA drives proliferation, leading to hyperplasia. Tumors progressively develop, and metastases form in ≤35% of mice that develop cSCC. Malignancy is influenced by mouse age and genetic background as well as the dose of initiator and the frequency of promoter application [57, 58]. In H-Ras null mice, DMBA/TPA treatment instead triggers K-Ras mutation, which results in 6-fold fewer papillomas but 4-fold more metastasizing cSCC [52, 59]. Although the DMBA/TPA model is invaluable to study cSCC, it must be noted that human skin responds to TPA differently than mouse skin. In another popular cSCC model, H-Ras mutation cooperates with CDK4 overexpression, which promotes G1 escape, to induce invasive cSCC from the xenografted human keratinocytes [60]. Age-associated inflammation promotes H-Ras mutation-driven tumorigenesis. When an inducible, basal, H-RasG12V construct was induced in young versus old mice, the young mice developed dysplasia while aged mice developed greater dysplasia with hyperplasia, inflammation, and marked macrophage and T-cell infiltration, ultimately developing cSCC. For young mice, dysplasia resolved upon 4-OHT withdrawal while aged epidermis remained hyperplastic and inflamed [61]. These studies highlight the importance of context in carcinogenesis, even when testing the same oncogene.
Although vital for Ras function, the GEFs remain understudied in epidermal development and tumorigenesis. While SOS1 is required for epidermal development, SOS2 is dispensable. SOS1 knockout in mice decreases keratinocyte proliferation, wound healing, and the number of epidermal and dermal layers and delays cSCC formation. In contrast, SOS2 knockout causes no change to skin architecture or tumor formation [62]. RasGRP1 is overexpressed in cSCC, yet RasGRP1 overexpression in normal keratinocytes induces G2 arrest. Like oncogenic H-Ras, RasGRP1 overexpression alone inhibits epidermal stratification in organotypic culture, but cooperates with mutant p53 to transform keratinocytes [63]. The limited evidence suggest that the role of Ras associated proteins in cSCC deserves more thorough investigation.
RAF and MEK. Raf proteins are serine/threonine kinases directly activated by Ras. Three Raf genes are encoded by the human genome: a-Raf, b-Raf, and c-Raf [64]. b-Raf mutations are highly prevalent in melanoma but rare in cSCC. Intriguingly, b-Raf inhibition, used to treat melanoma, induces Ras signaling and cSCC in a subset of patients (Fig. 3) [65, 66]. These cSCC tend to occur in sun-protected skin, and 60% harbor Ras mutation [66]. In the DMBA/TPA mouse model, b-Raf inhibition accelerates tumor formation without increasing tumor number [67]. In the presence of K/N-Ras mutation, b-Raf inhibitors drive b/c-Raf heterodimerization activating c-Raf, MEK, and ERK [67, 68]. In this context, MEK inhibition does not affect established tumors, but suppresses new tumor formation by 91% [67]. These findings reveal complex regulation among the different modules of the Ras pathway.
MEK1 and MEK2 kinases are downstream of Raf. MEK1 and MEK2 are individually dispensable for epidermal development while double knockout induces severe proliferation and barrier defects and rapid post-natal lethality. Downstream ERK2 activation rescues this hypoplasia [69]. Activated Raf can utilize either MEK1 or MEK2 to induce hyperplasia [69]. However, overexpression of either wild-type or kinase dead MEK1, but not MEK2, induces proliferation and reversible epidermal hyperplasia [70], suggesting that MEK1 upregulation differs from MEK2 in contributing to cSCC progression. MEK inhibitors were reported to cause senescence of cSCC cells in vitro and abrogate tumor development in UV-induced cSCC mouse models [64]. MEK inhibitors may be a valuable cSCC therapeutic option, but toxicity must still be carefully evaluated.
NOTCH Pathway
The NOTCH pathway, essential for both embryonic development and epidermal homeostasis, also plays a crucial role in cSCC progression. Four membrane bound NOTCH receptors are encoded by the human genome but only NOTCH1, 2, and 3 are expressed in keratinocytes. Upon activation by a battery of ligands, the NOTCH intracellular domain translocates to the nucleus to regulate transcription. NOTCH is frequently mutated in cSCC: NOTCH1 42.6% to 75%, NOTCH2 18% to 59%, and NOTCH3 7.4% [7, 44, 55, 71, 72]. While the role of NOTCH in cancer appears to be tissue specific, NOTCH1 generally functions as a tumor suppressor in the epidermis. NOTCH1 knockout induces epidermal hyperplasia, disrupts differentiation, and promotes tumor formation upon chemical induction [73, 74]. At the transcriptional level, MAML1 functions as a critical NOTCH coactivator. A dominant negative mutant of MAML1 inhibits NOTCH signaling and induces epidermal hyperplasia with dermal hypoplasia and lesions consistent with AK and cSCC. This hyperplastic tissue features cyclin D accumulation in the nuclei of suprabasal cells, suggesting suprabasal cells fail to exit the cell cycle during differentiation without NOTCH signaling [75]. Additionally, engineered pulses of NOTCH1 activation rapidly induce spinous differentiation and the DNA damage response, but induction of terminal differentiation is delayed. NOTCH1 activation also fails to downregulate basal markers, such as p63 [76]. Together these results show that NOTCH1 is necessary, but not sufficient for induction of epidermal differentiation. Yet, despite the described role as a differentiation inducer and tumor suppressor, NOTCH1 is overexpressed in CD133+ cells, which can initiate new tumor formation upon mouse xenograft. NOTCH1 silencing reduces the CD133+ population in cSCC cell lines by 40% and colony formation [77], suggesting that NOTCH1 may also play a role in cSCC initiation.
TGFβ Pathway
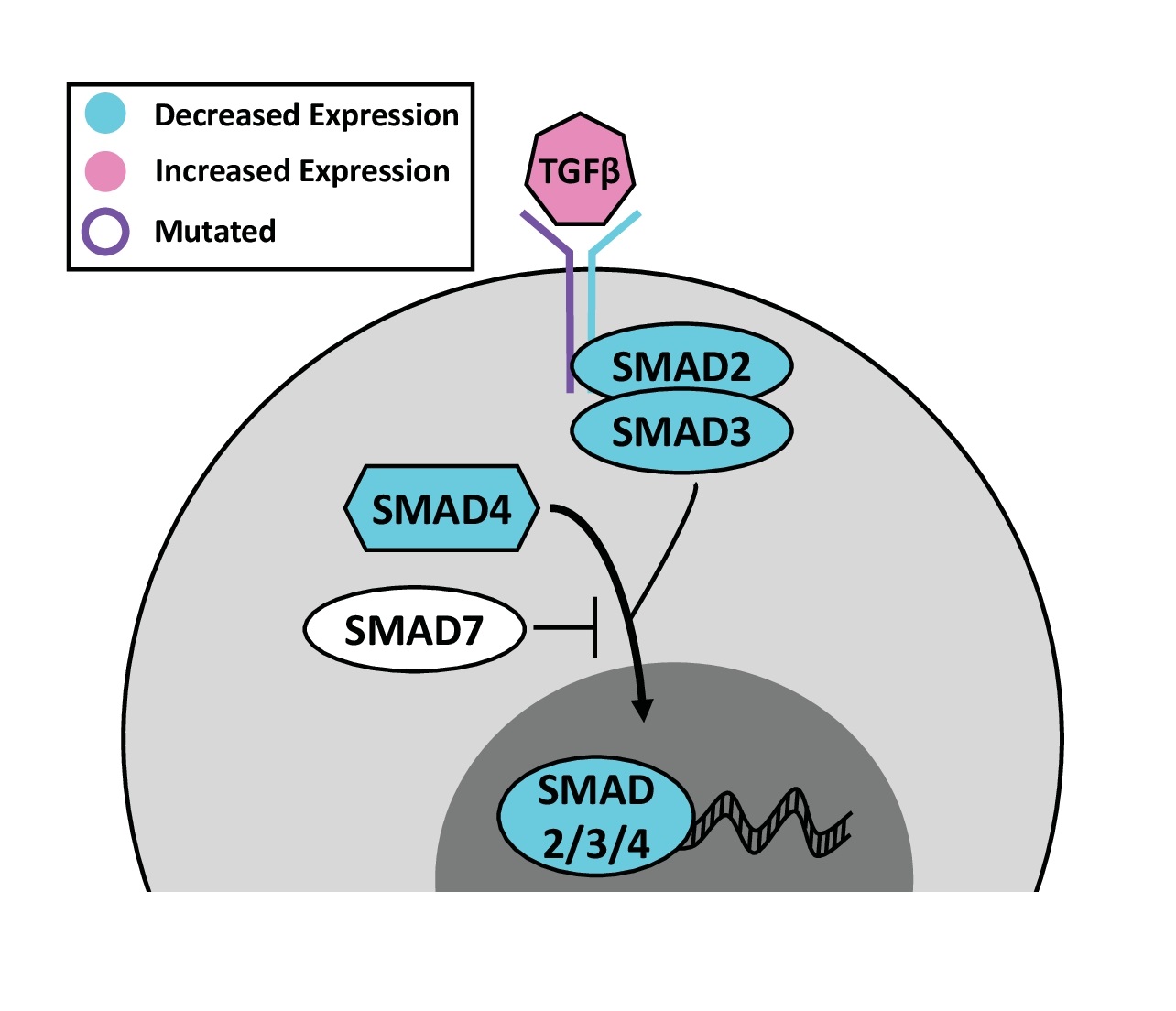
The TGFβ signaling pathway is comprised of 35 TGFβ ligands, 13 receptors, and eight SMAD effectors. For a detailed TGFβ review, see Wu et al. [78]. The role of TGFβ signaling changes as cSCC progresses. In early tumorigenesis, TGFβ signaling generally acts as a suppressor by reducing keratinocyte proliferation, but switches to an oncogenic role as cSCC advances. For example, TGFβ secreted by the stroma, slows cSCC cell cycling at the invasive front of the tumor, which helps to confer resistance to cisplatin chemotherapy. Consequently, these TGFβ responsive cells can induce tumor recurrence [79].
TGFβ ligand initiates signaling by activating a receptor complex of TGFBR1 and TGFBR2, which then phosphorylates SMADs 2 and 3. In cSCC, TGFBR1 and TGFBR2 are recurrently mutated (Fig. 4), primarily resulting in loss-of-function and inactivation of SMAD signaling. TGFBR2 mutant cSCC cell lines (SCCIC8, SCCIC12) fail to respond to TGFβ treatment and continue proliferating [80]. On the surface, this would suggest that SMAD signaling suppresses cSCC development, but the downstream SMAD proteins actually play opposing roles. Some studies indicate that SMADs function as suppressors for cSCC. For example, SMAD2 and SMAD4 are recurrently lost in cSCC tumor samples by IHC (Fig. 4). The reduction of phosphorylated SMAD2 and SMAD3 in human cSCC correlates to larger, thicker tumors [81]. Epidermal SMAD4 knockout induces spontaneous cSCC formation in 70% [82] to 100% [83] of mice within a year. Likewise, epidermal SMAD2 knockout promotes cSCC formation during chemical carcinogenesis [84]. In contrast, SMAD3 knockout dramatically reduces the number of papillomas and cSCC tumors that form in response to DMBA/TPA, suggesting that SMAD3 promotes cSCC progression [85]. In addition to regulating proliferation, TGFβ signaling can serve as a powerful monocyte chemoattractant. Mice overexpressing the inhibitory SMAD7 are neither more nor less susceptible to chemical carcinogens [86]. Interestingly both SMAD3 knockout and SMAD7 overexpression result in decreased tumor macrophage infiltration [85]. Altogether, the conflicting roles of the individual components make drugging the TGFβ pathway fraught with the danger of exacerbating cSCC.
Alterations to Effectors
p53
The most commonly mutated gene in cSCC is p53 with reported mutation rates of 64-85.2% [7, 44, 54]. p53 functions as the “guardian of the genome,” and suppresses cancer initiation by promoting cell cycle arrest, DNA repair, or apoptosis in response to metabolic disorder and DNA damage. Since p53 functions as a tetramer, the mutant protein can antagonize the remaining wild-type protein and can even abolish its transcription regulatory activity.
Mutant p53 resists protein degradation and accumulates in the nucleus, which enables IHC analyses of clinical samples [87]. IHC demonstrates that p53 mutation occurs early during epidermal carcinogenesis. In >90% of AK, more than half the cells are p53 positive. Positivity significantly correlates with cumulative UV exposure, older age, and high-grade dysplasia [88]. AK that persist demonstrate more intense p53 staining than AK that spontaneously regress [89]. p53 staining intensity further increases upon progression to cSCC, and high positivity delineates poorly differentiated from well-differentiated tumors [90]. In metastatic disease, p53 mutation is retained [71] and promotes aggressive tumor behavior such as recurrence [54], but additional p53 mutations are not usually acquired [91]. These studies demonstrate that p53 mutation promotes cSCC initiation and progression.
In addition to damaging genome stability, p53 mutations can cooperate with other oncogenes, such as Ras, to suppresses differentiation, promote proliferation, and induce large, poorly differentiated tumors that undergo epithelial to mesenchymal transition (EMT) [56]. Therefore, numerous therapeutics targeting p53 have been developed to restore its anti-cancer functions. MDM2 functions as the primary inhibitor of p53 by marking it for proteasomal destruction. Eight inhibitors of the MDM2-p53 interaction are undergoing phase I and II clinical trials. In other solid tumors and leukemias harboring wild-type p53, relieving MDM2 inhibition holds promise for treating malignancy. However, only a minority of cSCC preserve wild-type p53. In addition, small molecules that can fold mutant p53 into the wild-type conformation are being developed, but only two compounds, APR-246 and COTI-2, have reached clinical trials [92, 93]. It remains unclear whether these compounds target specific mutations or can broadly refold mutant p53. Another therapeutic strategy reduces mutant p53 protein accumulation. One such therapy, ganetespib, is a Hsp90 inhibitor that also reduces p53 accumulation, but a phase III clinical trial for advanced non-small cell lung cancer failed to show improved survival [94]. The promise of p53 therapies remains untested in cSCC. However, it will be critical that these p53 drugs, especially the refolding agents, do not also inadvertently stabilize the homologous p63 protein, a key cSCC driver. If so, p53 therapies could instead promote cSCC growth.
p63
p63 is a p53 paralog and serves as the master regulator of epidermal development and homeostasis. The p63 locus produces different isoforms including TA (has p53-like transactivation domain) and ΔN (lacks transactivation domain). p63 deletion in mice induces a lethal absence of epidermis due to dysregulation of both proliferation and differentiation [95]. ΔNp63α is the predominant functional isoform in the epidermis, where it promotes basal keratinocyte proliferation through p53 suppression while simultaneously controlling differentiation through a p53-independent mechanism [96].
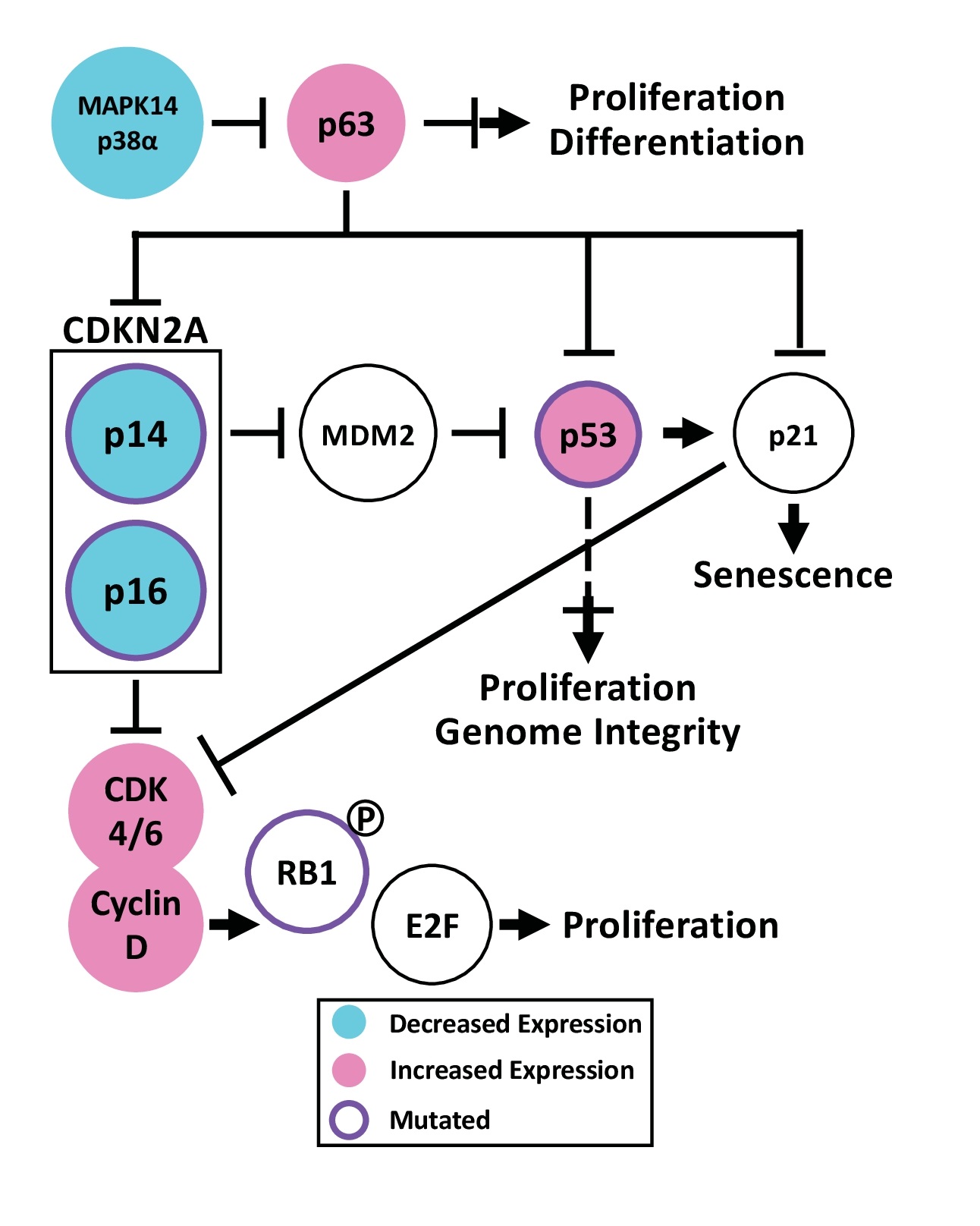
Sequencing of cSCC tumors reveals recurrent p63 amplification [71], but not mutation (Fig. 5). Consistent with sequencing, IHC demonstrates significantly increased p63 protein in cSCC [97]. In vivo experiments using mouse models demonstrate that ΔNp63α overexpression promotes cancer progression. p63 overexpression impairs apoptosis after UV exposure [98] and induces epidermal hyperplasia without spontaneous tumor formation. During DMBA/TPA tumor induction, p63 overexpression accelerates and increases tumor formation [99]. Consistently, factors enhancing p63 stability also promote carcinogenesis. STXBP4 opposes p63 degradation mediated by the anaphase promoting complex. In human cSCC, high STXBP4 expression significantly correlates with high ΔNp63α expression and poor disease staging. In a mouse xenograft model, overexpression of p63, degradation-resistant p63, or STXBP4 increases tumor volume [100]. Similar to the anaphase promoting complex, p38α (MAPK14) marks p63 for proteasomal destruction. While AK retain some p38α, it is dramatically reduced in cSCC (Fig. 5). Mice with epidermal p38α knockout express greater p63 protein causing increased keratinocyte proliferation and migration. During DMBA/TPA tumor induction, p38α knockout mice develop more papillomas than WT mice with unique malignant features including: full-thickness proliferation, impaired differentiation, vascularization, and myeloid infiltration [101]. Interestingly, ΔNp63α induces expression of MKP3, a phosphatase that represses p38 activity. ΔNp63α knockdown increases p38 phosphorylation, leading to p21 phosphorylation, Rb dephosphorylation, and G1 arrest [102]. These studies demonstrate that mechanisms increasing the amount of p63 protein can promote cSCC.
ΔNp63α represses CDKN2A, which produces the two tumor suppressors, p16 (p16INK4a) and p14 (p14Arf). In cultured keratinocytes, ΔNp63α overexpression bypasses passage-induced senescence through p14/p16 inhibition [99, 103]. p16 loss selectively immortalizes keratinocytes by increasing telomere length and maintaining expression of hyperphosphorylated Rb. Despite immortalization, the cells can stratify and differentiate with prominent hyperproliferation [104]. Further, p63 overexpression and p14 knockout can cooperate in carcinogenesis. p63 overexpression increases expression of basal keratinocyte markers, but these cells can initiate differentiation yet continue to proliferate abnormally [56]. Upon xenograft, ΔNp63α overexpressing or p14 knockout keratinocytes form poorly differentiated, necrotic tumors [56, 103] while combining ΔNp63α overexpression with p14 knockout significantly increases tumor volume [103].
In contrast to ΔNp63α, the TAp63 isoform functions as a tumor suppressor. TAp63 is not normally detectable in epidermis but is induced upon stress [105]. A component of the Fanconi anemia pathway, FANCD2 stimulates TAp63 expression in response to DNA damage [106]. TAp63 knockout induces DNA damage, including chromosome aberrations and aneuploidy. Knockout mice chronically exposed to UVR develop more primary cSCC tumors and subsequent lung metastases [107]. Finally, TAp63 is required for oncogene-induced senescence [106].
p73
The other p53 paralog, p73 is expressed in basal keratinocytes but is dispensable for epidermal development and differentiation. Knockout mice demonstrate impaired wound healing due to increased DNA damage and decreased proliferation at the wound edge [108]. In various cancers, p73 on chromosome 1p36 is frequently lost, but rarely mutated [109]. Cultured cells treated with DMBA lose p73 protein during malignant conversion. Additionally, TAp73 knockdown keratinocytes resist radiation-induced apoptosis and senescence and form malignant tumors upon xenograft. Restoration of TAp73, but not ΔNp73, reduces tumor formation [110]. Compound p53+/- p73+/- heterozygous mice spontaneously develop cSCC that readily metastasize [111]. Overall, these limited results suggest that p73, especially TAp73, functions as an epidermal tumor suppressor of cSCC.
Dysregulation of the G1/S Checkpoint
Uncontrolled proliferation is a hallmark of cancer. The “restriction point,” also known as the R point, serves as a critical checkpoint for cells to incorporate environmental cues before entering the cell cycle. R point regulation is often disrupted in cancer. Regulation of retinoblastoma (Rb) phosphorylation controls the R point, before G1 to S transition. The cyclin D1-CDK4/6 complex initiates Rb phosphorylation and the release of pro-proliferative E2F transcription factors. In cSCC, CDK4/6, cyclin D1, and Rb are frequently dysregulated.
Overexpression of cyclin D1 in keratinocytes increases proliferation, even in the presence of increased calcium, which normally induces terminal differentiation. While normal skin is typically negative for cyclin D1 by IHC, BD and cSCC (70-87% positive) demonstrate progressively increasing positivity, which correlates to sun exposure and worsening dysplasia [112-115]. At least 20% of cSCC exhibit gain of chromosome 11q13, where cyclin D1 resides (Fig. 5) [116]. In mice, oncogenic H-Ras increases cyclin D1 expression, cyclin D1-CDK4 association, and Rb phosphorylation. Cyclin D1 knockout delayed tumor formation and reduced tumor size and multiplicity upon TPA/DMBA treatment [117]. DMBA treatment induces faster and increased papilloma formation in mice overexpressing cyclin D1 [118]. Thus, increased cyclin D1 expression is a recurrent feature associated with cSCC.
Like cyclin D1, CDK4/6 is upregulated in cSCC tumors (Fig. 5), in part from the reduction of the transcription factor NFIB, which normally represses their expression. NFIB knockdown induces larger tumors with increased CDK4/6 and pRb [119, 120]. The CDK4/6 inhibitor, rafoxanide, decreases total and phosphorylated CDK4/6, cyclin D1, and pRB thus suppressing the G1/S transition and reducing cSCC tumor volume in mice [121].
Downstream, RB1 is mutated in 11.5 to 18.2% of cSCC tumors (Fig. 5) with an enrichment of nonsense and splice site mutations [44, 122]. While Rb is dispensable for epidermal development, knockout induces a lifetime of epidermal hyperplasia without spontaneous tumor formation despite its well-known tumor suppressor function. Mice with epidermal knockout of both Rb and E2F1 exhibit enhanced hyperplasia and disordered differentiation and develop wound-like cSCC tumors, indicating that E2F1 loss promotes cSCC in the absence of Rb [123]. Epidermis requires intact Rb and E2F1 function to maintain homeostasis and to suppress cSCC formation.
CDKN2A produces p14 and p16, two distinct tumor suppressors through alternative splicing. p16 binds CDK4/6 to inhibit Rb phosphorylation while p14 stabilizes p53 by inhibiting MDM2. cSCC demonstrates pervasive CDKN2A inactivation through a variety of mechanisms, including recurrent loss of chromosome 9p21 [71, 122], promoter hypermethylation [124], and mutation affecting 23% [7] to 61.5% of tumors (Fig. 5) [44, 55]. These alterations correlate with disease-specific death and shorter survival [91]. In mice, CDKN2A knockout enhances UV-induced tumor formation [125]. Given their importance, mutant p14/16 are attractive targets, but are considered difficult to drug. Epigenetic alterations to CDKN2A are being targeted by drugs such as 5-Aza-2’-dexoycytidine, approved for hematological malignancies, to induce CDKN2A promoter demethylation [126]. However, this approach cannot address CDKN2A mutation.
MYC
MYC is essential for epidermal homeostasis and wound healing. Embryonic c-MYC ablation reduces basal cellularity and induces premature differentiation causing skin tightness that limits movement and slow-healing wounds in adult mice [127]. Mice with conditional, epidermal c-MYC ablation resist Ras-induced oncogenesis [128]. In contrast, sustained suprabasal expression of c-MYC induces proliferation, hyperplasia, papilloma formation, and angiogenesis [129], which are key processes associated with cancer progression.
AK and cSCC frequently demonstrate MYC positivity by IHC, which associates with poor tumor differentiation [130, 131]. Increased MYC expression in cSCC results from genomic amplification [71] and post-translational modifications that increase MYC protein stability. Serine 62 phosphorylation, which increases stability, is increased, and threonine 58 phosphorylation, which targets MYC for degradation, is consistently reduced in patient cSCC tumors. During DMBA/TPA treatment, epidermal expression of MYCT58A (a degradation resistant mutant) decreases tumor latency and increases malignant transformation and metastasis compared to MYC overexpression. MYC overexpression actually decreases the epidermal stem cell population while MYCT58A increases stem cell number and drives the stemness program [132]. As in other cancers, MYC functions as a potent oncogene in epidermal tissue.
Epigenetic regulators
Epigenetic regulators, including histone/DNA modifiers and ATP-dependent chromatin remodelers, play critical roles in gene regulation. For an overview of this topic, please refer to Audia et al. [133]. These regulators and the epigenetic landscape are commonly dysregulated in cancer, including cSCC. Here, we present the dysregulation of methylation, acetylation, and chromatin remodelers described in cSCC.
DNA Methylation. DNA methylation dynamics in normal epidermis are altered with age and sun exposure. Analysis of sun exposed and protected skin from young and old individuals reveals that dermal methylation remains unaffected while the epidermis experiences dramatic methylation changes influenced by both aging and sun exposure. Widespread heterochromatin hypomethylation in aged and exposed samples affects the same sites hypomethylated in cSCC [134]. In a survey of sun-exposed/protected skin samples, methylation readers and writers were nearly universally dysregulated in sun exposed skin with increased DNA methyltransferases (DNMT1 and DNMT3B) and decreased DNMT3A and the demethylases TET1, TET2, and TET3 [135]. Analysis of AK, primary cSCC, and metastatic cSCC reveals that CpG methylation decreases during the transition from AK to cSCC and then increases in metastatic cSCC. Gene bodies gain methylation while promoters preferentially lose methylation [136]. When the epidermal portion of cSCC and normal skin is separated and subjected to bisulfate sequencing, promoter and gene bodies demonstrate hypermethylation while repetitive sequences upstream of and within gene bodies are hypomethylated [135]. The methylation profiles of normal skin are relatively homogeneous when compared to the heterogenous methylation displayed by AK and cSCC. However, AK and cSCC consistently cluster into two groups, suggesting that some lesions arise from more progenitor-like precursors while others arise from more differentiated cells [137].
Histone Methylation. The enzymes controlling histone methylation are also frequently dysregulated in cSCC. KDM1A is a repressive histone demethylase that removes H3K4me. KMD1A is overexpressed [138] while the opposing lysine methyltransferases KMT2C and KMT2D are mutated in 39% and 69.2% of cSCC respectively [72]. KMT2C mutation significantly associates with invasion and decreased survival [72]. These modifiers may represent therapeutic targets since KDM1 inhibition induces differentiation and reduces dermal invasion in mice [138]. The polycomb repressive complexes also write histone methylation. RING1B of complex 1 and EZH2 of complex 2 are increased in metastasizing versus non-metastasizing cSCC [139]. EHMT2, an H3 lysine methyltransferase, is increased in cancer. EHMT2 deletion in mice increases chromatin accessibility at regulatory sites with minimal gene expression changes. However, deletion induces replicative stress and genomic instability. While knockout delays cSCC induction, the tumors are aggressive. Depletion in established tumors causes regression, but regressed lesions relapse into more aggressive, poorly differentiated tumors [140]. Thus, this EHMT2 study provides a cautionary tale about the unexpected outcomes of genome-wide modification.
Acetylation. The evidence regarding the role of CREBBP and p300 in cSCC is limited and conflicting. Both are lysine acetyltransferases that function as transcriptional activators. At least one functional allele of each is required for epidermal development. CREBBP mutation affects ~30% of cSCC tumors. In contrast to other cancers, which disclose indel mutations, primary cSCC tumors harbor CREBBP missense mutations. Yet, cSCC lymph node metastases acquire CREBBP indels, suggesting loss of function promotes tumor progression [141]. In contrast, increased expression of p300 significantly associates with lymph node metastasis, advanced tumor stage, and poor survival [142, 143]. In mice, heterozygous loss of EP300 or CREBBP cooperates with HRASS35 to induce postnatal epidermal thickening and spontaneous papilloma formation due to EGFR hyperactivation [144].
Preclinical testing in murine cSCC models suggests that HDAC inhibitors could be a promising cSCC treatment strategy. HDACs are histone deacetylases, and HDAC inhibitors are already approved for a number of hematological malignancies. HDAC3 is increased in cSCC. The ginseng derived compound 20(R)-Rg3 decreases HDAC3 protein. Either drug treatment or HDAC3 knockdown reduces EMT and migration [145]. Likewise, the compound MS-275 inhibits class I HDACs leading to upregulation of H3K9ac. Low-dose treatment inhibits the proliferation of human cSCC cell lines and significantly reduces the size of UV-induced tumors [146]. Yet another inhibitor, vorinostat reduces HDAC1/2/3/7 activity and increases histone and non-histone acetylation. Treatment reduces proliferation, increases apoptosis, and induces large areas of necrosis in cSCC xenografts [147]. Targeting cSCC acetylation appears promising, and FDA-approved drugs can be repurposed more quickly than new drugs can be developed.
The BAF Complex. The BAF family of chromatin remodeling complexes regulate DNA accessibility for transcription. BAF genes are highly mutated in cancer and can function either as an oncogene or tumor suppressor in a tissue dependent manner [148]. The ATPase subunits, BRM and BRG1 are significantly and consistently reduced in cSCC tumors by
IHC [149]. BRM knockout induces severe epidermal hyperplasia and abnormal proliferation of the suprabasal keratinocytes in mice upon UV radiation [150]. The BRM null cells enter G1 arrest but escape prematurely and accumulate DNA damage without appropriate DNA repair or apoptosis [151]. Another BAF subunit, ACTL6A, suppresses the differentiation program in progenitors by sequestering the complex from promoters of genes required for differentiation. Consequently, ACTL6A loss induces cell cycle exit and epidermal hypoplasia [152]. cSCC exhibits intense ACTL6A overexpression, and 20% of tumors harbor ACTL6A amplifications. Unlike in normal epidermis, ACTL6A and p63 are co-expressed in cSCC and robustly interact to promote the progenitor transcriptional program. ACTL6A knockdown inhibits proliferation, invasion, and xenograft tumor growth while overexpression promotes rapid growth [153, 154]. Within the same complex, BRM/BRG1 and ACTL6A play opposing roles in cSCC formation. Chromatin remodelers remain understudied in epidermal homeostasis and cSCC despite containing potentially druggable ATPase domains.
Alterations to Non-coding RNAs
An emerging literature indicates that non-coding RNAs (ncRNAs) play crucial roles in regulating epidermal differentiation, wound healing, and UV response and function as both oncogenes and suppressors in cSCC [155]. The ncRNAs characterized in cSCC include long non-coding RNAs (lncRNAs), microRNAs and circular RNAs, summarized in Table 1. These ncRNAs can influence gene expression in both the nuclear and cytoplasmic compartments. However, relatively few of these ncRNAs have been thoroughly characterized in normal epidermal development or homeostasis.
lncRNAs are transcripts >200 nucleotides with diverse regulatory functions including: scaffolding molecular complexes, guiding the binding of chromatin remodelers and transcription factors, modulating mRNA processing, and inhibiting (sponging) microRNAs [156]. Comparing cSCC to healthy skin by RNA sequencing reveals differential expression of 908 lncRNAs in cSCC with a bias towards downregulation [157, 158]. circRNAs are subset of lncRNAs with covalently closed ends [156], which share the downregulated trend (53/55 decreased) in cSCC. Additionally, the negative regulator of circRNA biogenesis, ADAR, is upregulated while positive regulators, MBNL and ESRP1, are downregulated [158]. All four of the circRNAs described in cSCC sponge microRNAs with tumor suppressor properties.
microRNAs are ~22 nucleotide RNAs that inhibit the translation of >60% of all mRNAs. Many of the microRNAs described in cSCC converge on the p63 and MAPK pathways, which are critical for epidermal homeostasis and carcinogenesis. microRNAs show therapeutic promise in preclinical models. For example, miR-634 synergizes with EGFR inhibition to reduce tumor growth in mice [159] while restoring miR-3619-5p expression in cisplatin resistant cell lines improves chemotherapy efficacy [160]. microRNA-based therapies are currently being developed and tested in clinical trials. Therapy is complicated by the potential for off-target effects and the myriad technical details regarding delivery of these fragile RNAs or their antisense inhibitors to tumors. For a summary of systemic delivery options, see Rupaimoole et al. [161] and Ross [162] for topical delivery.
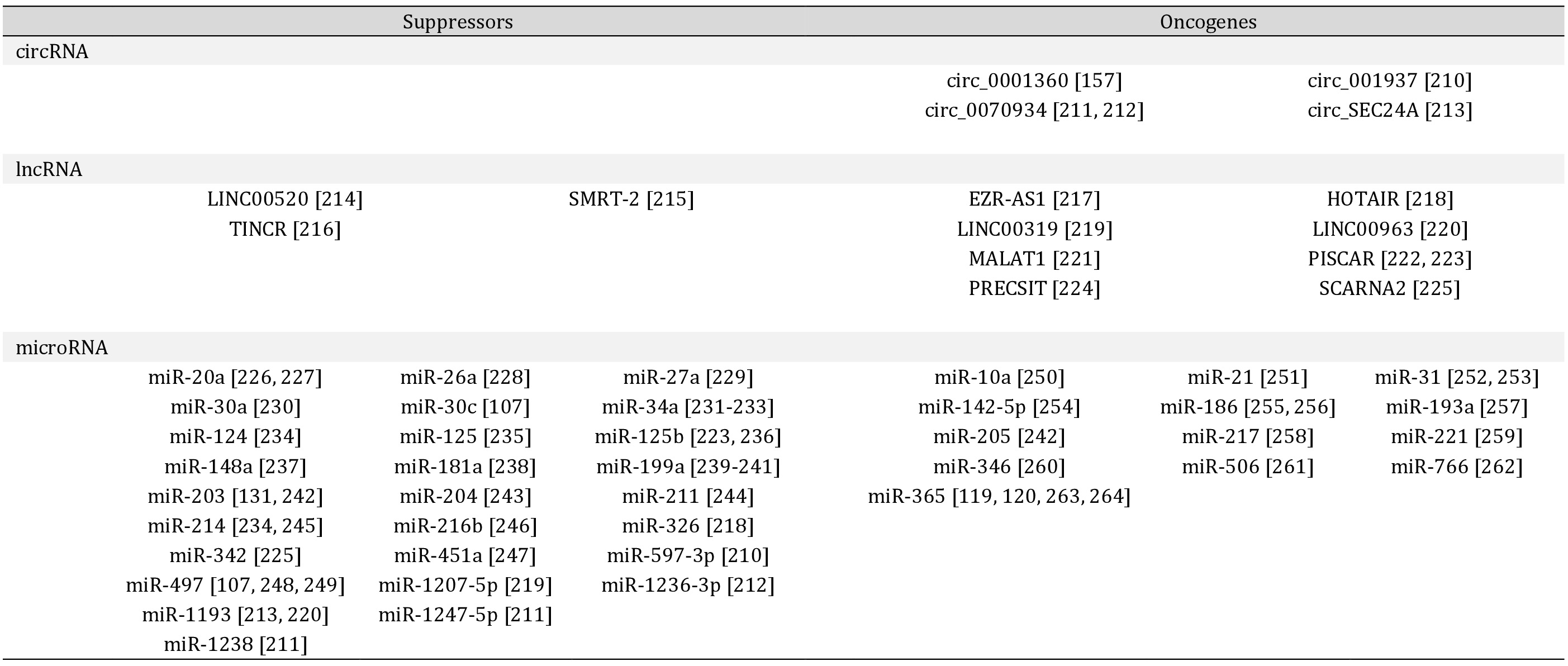
A summary of non-coding RNAs and their described roles as tumor suppressors or oncogenes in cSCC
Alterations to the Extracellular Environment
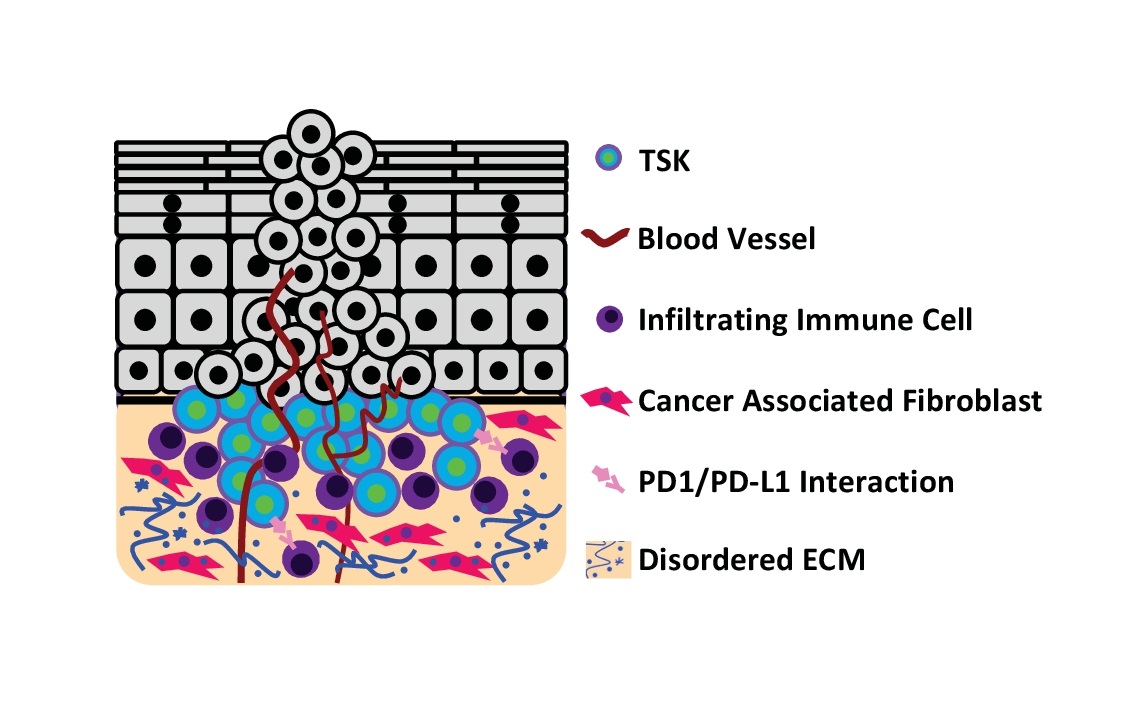
cSCC cells at the invasive front interact with the various components of the dermis, including: dermal fibroblasts, immune cells, and the extracellular matrix (ECM). Dermal fibroblasts secrete and organize the ECM, which is primarily composed of proteins and carbohydrates that orient and anchor cells and participate in signaling. Fibroblasts and immune cells secrete enzymes that digest the ECM, which contributes to angiogenesis and cSCC invasion. Finally, immune surveillance is perturbed. This section provides an overview of these alterations (Fig. 6) and their contribution to cSCC progression.
Dermal Fibroblasts
cSCC alters fibroblast gene expression and behavior, producing a population of cancer associated fibroblasts (CAFs). These CAFs cross talk with keratinocytes to promote cSCC progression. Ras-mutant HaCaT cells cannot invade collagen gels unless fibroblasts are present [163]. Further, cSCC cell lines cultured with primary CAFs in organotypic culture detach from and invade the dermis. Co-culture with CAFs impairs epidermal differentiation, but paradoxically reduces cSCC proliferation compared to co-culture with normal fibroblasts [164]. In mice, fibroblast proliferation increases during DMBA/TPA induction. Fibroblast depletion delays papilloma formation, reduces malignant transformation by half, and reduces macrophage tumor infiltration [165].
CAFs isolated from patient tumors demonstrate dysregulation of key regulatory pathways. CAFs associated with AK and cSCC demonstrate downregulation of ATF3, a transcription factor transiently induced by UV radiation in normal fibroblasts. ATF3 deletion in fibroblasts promotes the secretion of growth factors, cytokines, and matrix modulating enzymes. Xenograft of ATF3 depleted fibroblasts with cSCC cells increases dysplasia and induces formation of invasive, ulcerated cSCC with impaired differentiation [166]. CAFs also demonstrate altered WNT and NOTCH signaling. About 25% of cSCC disclose nuclear beta-catenin in CAFs instead of the membranous staining observed in normal dermis. These CAFs are highly susceptible to WNT induction, which induces secretion of cytokines and matrix components promoting keratinocyte proliferation [167]. Further, CAFs isolated from patient cSCC tumors recurrently disclose NOTCH1 amplification. Silencing NOTCH1 partially normalizes CAF gene expression, impairs cSCC cell growth in co-culture, and reduces tumor size, macrophage infiltration, and angiogenesis in xenograft [168]. Additionally, CAFs exhibit dysregulated FGF and TGFβ signaling, which oppositely regulate the transcription factor ETV1. Abnormal signaling increases growth factor and cytokine production, ECM remodeling, macrophage recruitment, and promotes tumor growth [169]. Both epidermal keratinocytes and dermal fibroblasts accumulate mutations and signaling alterations which drive abnormal cellular interactions and cSCC progression.
Tumor Infiltrating Immune Cells
Homeostatic skin hosts a battery of resident immune cells [170]. During malignant transformation, additional immune cells are recruited, where they contribute to cancer progression. For example, DMBA/TPA induction recruits neutrophils. In papillomas, neutrophils promote ECM remodeling, angiogenesis, and metastasis while neutrophils in tumors alter metabolism and suppress immune response [171]. Xeroderma pigmentosum, caused by a deficiency in DNA repair, promotes childhood cSCC development. Fibroblasts from both xeroderma pigmentosum patients and sporadic cSCC lose the receptor CLEC2A, which activates natural killer cells. In organotypic culture containing cSCC cell lines, normal fibroblasts, and natural killer cells, introduction of anti-CLEC2A antibody promotes invasion to the same extent as culture containing xeroderma pigmentosum fibroblasts [172]. Compounding natural killer inactivation, greater than 75% of spontaneous cSCC tumors express another cell-surface receptor LLT1, which inhibits natural killer cell. Expression of LLT1 associates with increased tumor thickness, nodal metastasis, and death [173]. Although neutrophils and natural killer cells are recruited to cSCC tumors, their normal anti-tumor activity is subverted or inhibited.
Although macrophages are the most abundant immune cell in homeostatic skin, premalignant and cSCC lesions progressively recruit even more macrophages [174]. These tumor associated macrophages (TAMs) primarily surround rather than infiltrate cSCC tumors and become dysfunctional. For example, normal macrophages can be broadly classified as either M1 (pro-inflammatory) or M2 (regulatory, wound healing) but many TAMs exhibit abnormal M1/M2 bi-activation [175]. Additionally, TAMs secrete VEGFC, MMP9, and MMP11 which collectively promote new vessel formation and metastasis [175, 176]. Because of their abundance and dysfunction, TAM depletion may be a viable strategy to treat cSCC. In a murine model, macrophage depletion inhibits tumor growth, decreases vessel penetration into tumor, and reduces invasion [177]. Likewise, a phase III trial demonstrates that oral nicotinamide dramatically reduces TAMs, which may contribute to its ability to prevent new tumor formation [178].
T cell dysfunction is a key feature of cSCC. T cells accumulate in the peritumor environment and express markers of exhaustion, which is the inability to effectively respond to antigen stimulation [13]. Dendritic cells process and present antigens to T cells. Like other immune cells, dendritic cells are increased in the peritumoral area. Yet, dendritic cells isolated from cSCC are unable to stimulate T cells compared to dendritic cells isolated from normal human skin [179]. In fact, cSCC-derived dendritic cells can even suppress T cell proliferation in ex vivo culture [180]. However, some cSCC associated immune cells increase T cell activation. Langerhans cells are epidermis-resident macrophages with dendritic properties. cSCC-associated Langerhans cells can induce greater proliferation of CD4+ and CD8+ T cells than Langerhans cells from normal skin. This suggests that utilizing tumor Langerhans cells in therapy could be a promising strategy [180].
Further contributing to T cell dysfunction, is the dysregulation of the PD-1/PD-L1 axis in cSCC. In healthy tissue, the programmed death-1 receptor (PD-1) and its ligands, PD-L1 and PD-L2, protect tissue by preventing excessive T cell activity. Tumor cells activate PD-L1 expression to escape T cell mediated killing. Additionally, tumor infiltrating immune cells also express PD-L1. This induces an immune suppressive phenotype in regulatory T cells, inhibits naïve CD8+ T cell activation, and prevents reactivation of exhausted CD8+ T cells, the cells that likely recognize tumor neoantigens [181].
The presence of PD-L1+ cells predicts lymph node metastasis and poor differentiation [182, 183]. However, disagreement remains in defining what constitutes PD-L1 positivity with different studies setting widely varying thresholds and examining different cell types. 26% of primary cSCC tumors from immunocompetent patients exhibit PD-L1+ tumor cells, 60% of cSCC harbor PD-L1+ infiltrating immune cells, and 81% have PD-1+/CD8+ T cells [184]. Another study showed that 41% of cSCC tumors harbor PD-L1+ dendritic cells and 61% contain PD-L2+ dendritic cells, which correlates with increased tumor size, and counterintuitively well-differentiated status [185]. During DMBA/TPA induction, PD-L1+ neutrophils infiltrate tumors and inhibit CD8+ T cells. Neutrophil depletion delays tumor growth [171]. Positivity persists in metastatic cSCC, where ~33% of tumor cells are PD-L1+ and correlates with intra- and peritumoral CD8+ T cell infiltration and poor differentiation of the primary tumor [186, 187]. Together these results suggest the PD-1 and PD-L1 positivity are prevalent in cSCC and that PD-L1 positivity implies poorer disease outcomes. In contrast, the role of PD-L2 has not been satisfactorily elucidated.
In 2018, the FDA approved cemiplimab, a PD-1 antibody, as the first treatment for advanced cSCC. The phase I trial enrolled 26 patients and achieved an overall response rate (ORR) of 50% while the phase II trial enrolled 193 immunocompetent patients and achieved ORR of 41.1-49.1% with durable response. Interestingly, responders had a higher mutational burden of 53.2 to 74.2 per Mb compared to 13.7 to 28.7 per Mb for non-responders, suggesting that mutational burden may predict patient response [188]. Likewise, pembrolizumab, another PD-1 antibody achieved an ORR of 34.2% in patients with unresectable cSCC [189]. cSCC tumors were classified as PD-L1 positive or negative by IHC. The ORR for PD-L1+ patients was 55% versus 17% for PD-L1- patients [190]. Currently, clinical trials are enrolling cSCC patients to test nivolumab (PD-1 antibody), IBI318 (PD-1/PD-L1 bispecific antibody), and avelumab, atezolizumab, and cosibelimab (PD-L1 antibodies). Immune checkpoint blockade will likely become standard of care for cSCC, which highlights the need for standardized biomarkers that predict patient response. Furthermore, managing toxicity remains critical, especially for organ transplant patients who risk graft rejection [191].
Extracellular Matrix
The extracellular matrix (ECM) is the non-cellular component of tissues which provides physical and biochemical support to the cells. Both the structural proteins and the enzymes that remodel them are dysregulated in cSCC. The chief structural ECM proteins are collagens. COL7A1 mutation causes recessive dystrophic epidermolysis bullosa (RDEB). Patients experience chronic blisters, fibrosis, inflammation, and early onset cSCC that readily metastasizes. The loss of COL7A1 induces disorganization of collagen fibers and fibroblast activation similar to that observed in spontaneous cSCC. In RDEB, the collagen cross-linker LOX, integrin β1, and the kinase FAK also increase which stiffen the ECM. This stiffening mechanically induces the pro-migration and pro-survival integrin-AKT signaling axis [192, 193]. cSCC tumors also demonstrate suprabasal stiffening. This combined with proliferation-driven tissue deformation and dysregulated remodeling of the basement membrane promotes invasive properties [194].
Integrins are heterodimeric cell surface receptors that mediate signaling to regulate cellular adhesion, migration, proliferation, and survival. Integrins are extensively dysregulated in cancer [195]. In the Ras-induced cSCC model, the stroma demonstrates rapid changes which promote angiogenesis followed by dysregulation of ECM and adhesion genes while antigen presentation is progressively repressed. Integrins (ITGB1, ITGB4, ITGA3, IGBA5, and ITGA6) govern this network of dysregulated stroma genes. ITGB1 increases during malignant progression, and antibody treatment reduces tumor growth and improves the epidermal-dermal boundary in a xenograft model [196]. Another integrin, ITGB4 is a component of the hemidesmosome, an anchor between keratinocytes and the ECM. ITGB4 undergoes increased N-glycosylation in cSCC. Interrupting this glycosylation reduces tumorigenesis [197]. Additionally, CAFs produce an integrin ligand, periostin. Although absent in normal ECM, periostin is prominently expressed in high risk cSCC and associates with larger tumors, invasion, and poor differentiation [198].
Laminins are heterotrimeric glycoproteins composed of α, β, and 𝛾 subunits. Laminin 332 (α3 β3 𝛾2) is the primary functional laminin in skin, and its receptors, integrins α3β1 and α6β4, are expressed on the surface of keratinocytes. Laminin α3 is decreased in poorly differentiated cSCC tumors [199]. IHC reveals 𝛾2 staining at the invasive edge of cSCC. Spheroid co-culture with fibroblasts induces Ras-mutant, but not wild-type, keratinocytes to produce lamin-332 and to invade collagen gels. Treatment with anti-integrin antibody prevents invasion in in vitro assays [200]. Knockdown of α3 or 𝛾2 reduces keratinocyte adhesion. In xenografts, α3 or 𝛾2 knockdown produces larger, more invasive tumors. β3 knockdown tumors are larger than control, but more differentiated and not invasive. α3 or 𝛾2 knockdown tumors recruit more macrophages that infiltrate the tumors [199].
Matrix metalloproteases (MMP) are calcium- and zinc-dependent enzymes that degrade components of the ECM. cSCC demonstrates an overall upregulation of MMP activity, which promotes invasion and angiogenesis. Tumor cells at the invasive front overexpress several MMPs (1, 2, 3, 7, 9, 11, 13, and 14). Invasive cells, but not normal epidermis, express MMP1, 3, and 7. In contrast, MMP9 and 10 are expressed by normal basal keratinocytes, but expression increases in cSCC [201-204]. Infiltrating immune cells contribute yet more MMP8 and 9 to the tumor environment [205]. cSCC and the adjacent dermis exhibit intense MMP10 staining, which associates with poor pathology [206]. MMP2, MMP14, and TIMP2 form a trimeric, membrane-bound complex. MMP14 activates MMP2. Weak MMP14 expression occurs in normal basal cells, and is elevated at the invasive front. MMP2 expression correlates with cSCC progression [201, 203, 207]. TIMP2 inhibits MMP2 and is reduced in cSCC tumors [201]. Additionally, the surface-bound glycoprotein CD147 stimulates MMP activity and is strongly expressed by primary cSCC and metastases. CD137 expression in primary tumors promotes metastasis [208]. Given the strong upregulation, MMPs are attractive therapeutic targets. Various MMP inhibitors are under pre-clinical and clinical investigation, but this approach is fraught with problems. Early, zinc-chelating inhibitors acted non-specifically and induced serious side effects. Antibody-based therapies provide specificity but are easily degraded. To achieve anti-tumor effects rather than inadvertently promoting metastasis, target selection, treatment timing, and drug specificity require further refinement [209].
Conclusion
cSCC represents an unmet clinical need that will continue to grow for the foreseeable future. cSCC develops from keratinocytes that accumulate a massive mutational burden, which impacts regulation of gene expression and signaling pathways. This mutational burden not only complicates the identification of important drivers of progression, but also facilitates synergy among oncogenes to accelerate cSCC progression. In addition, abnormal dermal fibroblasts and an abundance of infiltrating immune cells contribute to the cancerous phenotype by shaping the microenvironment. Understanding the complex interactions between these cellular components is crucial to fully describe cSCC dynamics and develop effective therapies. Currently, the only two approved therapies for cSCC are cetuximab, an anti-EGFR antibody that targets keratinocyte dysfunction, and cemiplimab, an anti-PD-1 antibody, that targets inappropriate immune interactions. Yet, both approved therapies would benefit from the discovery of biomarkers that robustly predict successful patient responses. Therapies specifically tested in and approved for cSCC are still needed. Fortunately, a number of promising targets have already been identified, and potential therapies are currently being developed or repurposed. Further research is required to discover novel druggable targets and biomarkers that reliably predict patient prognosis.
Author Contributions
SD and XB conceptualized the review. SD drafted manuscript, and XB advised on analysis and revision.
Funding
This work is supported by a NIH R01 (AR075015), an American Cancer Society Research Scholar Grant (RSG-21-018-01), Searle Leadership Fund, the Northwestern Skin Disease Research Center Pilot & Feasibility Award, the Basic Insights Award from Northwestern Cancer Center, and a NUCATS pilot award to XB, as well as a NIH/NCI F31 (F31CA261114) Fellowship to SD.
Statement of Ethics
The authors have no ethical conflicts to disclose.
The authors have no conflicts of interest to declare.
| 1 Tripathi R, Knusel KD, Ezaldein HH, Bordeaux JS, Scott JF: Characteristics of Patients Hospitalized for Cutaneous Squamous Cell Carcinoma. Dermatol Surg 2020;46:742-746. https://doi.org/10.1097/DSS.0000000000002117 |
||||
| 2 Que SKT, Zwald FO, Schmults CD: Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol 2018;78:237-247. https://doi.org/10.1016/j.jaad.2017.08.059 |
||||
| 3 Siegel RL, Miller KD, Jemal A: Cancer statistics, 2019. CA Cancer J Clin 2019;69:7-34. https://doi.org/10.3322/caac.21551 |
||||
| 4 Muzic JG, Schmitt AR, Wright AC, Alniemi DT, Zubair AS, Olazagasti Lourido JM, Sosa Seda IM, Weaver AL, Baum CL: Incidence and Trends of Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma: A Population-Based Study in Olmsted County, Minnesota, 2000 to 2010. Mayo Clin Proc 2017;92:890-898. https://doi.org/10.1016/j.mayocp.2017.02.015 |
||||
| 5 Fuchs E: Scratching the surface of skin development. Nature 2007;445:834-842. https://doi.org/10.1038/nature05659 |
||||
| 6 Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber DS, Roels S, White J, Otto GA, Ross JS, et al.: Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. https://doi.org/10.1186/s13073-017-0424-2 |
||||
| 7 South AP, Purdie KJ, Watt SA, Haldenby S, den Breems N, Dimon M, Arron ST, Kluk MJ, Aster JC, McHugh A, Xue DJ, Dayal JH, Robinson KS, Rizvi SH, Proby CM, Harwood CA, Leigh IM: NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol 2014;134:2630-2638. https://doi.org/10.1038/jid.2014.154 |
||||
| 8 Wang Y, Masaki T, Khan SG, Tamura D, Kuschal C, Rogers M, DiGiovanna JJ, Kraemer KH: Four-dimensional, dynamic mosaicism is a hallmark of normal human skin that permits mapping of the organization and patterning of human epidermis during terminal differentiation. PLoS One 2018;13:e0198011. https://doi.org/10.1371/journal.pone.0198011 |
||||
| 9 Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock D, Livitz D, Li X, Arich-Landkof E, Shoresh N, Stewart C, Segre AV, Branton PA, Polak P, Ardlie KG, Getz G: RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019;364:eaaw0726. https://doi.org/10.1126/science.aaw0726 |
||||
| 10 Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ: Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015;348:880-886. https://doi.org/10.1126/science.aaa6806 |
||||
| 11 Zilberg C, Lee MW, Yu B, Ashford B, Kraitsek S, Ranson M, Shannon K, Cowley M, Iyer NG, Palme CE, Ch'ng S, Low TH, O'Toole S, Clark JR, Gupta R: Analysis of clinically relevant somatic mutations in high-risk head and neck cutaneous squamous cell carcinoma. Mod Pathol 2018;31:275-287. https://doi.org/10.1038/modpathol.2017.128 |
||||
| 12 Patel GK, Yee CL, Terunuma A, Telford WG, Voong N, Yuspa SH, Vogel JC: Identification and characterization of tumor-initiating cells in human primary cutaneous squamous cell carcinoma. J Invest Dermatol 2012;132:401-409. https://doi.org/10.1038/jid.2011.317 |
||||
| 13 Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstrahle J, Larsson L, Bai Y, Zhu B, Bhaduri A, Meyers JM, Rovira-Clave X, Hollmig ST, Aasi SZ, Nolan GP, Lundeberg J, et al.: Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020;182:497-514 e422. https://doi.org/10.1016/j.cell.2020.05.039 |
||||
| 14 Mohandas P, Lowden M, Varma S: Bowen's disease. BMJ 2020;368:m813. https://doi.org/10.1136/bmj.m813 |
||||
| 15 Korhonen N, Ylitalo L, Luukkaala T, Itkonen J, Haihala H, Jernman J, Snellman E, Palve J: Premalignant lesions, basal cell carcinoma and melanoma in patients with cutaneous squamous cell carcinoma. Arch Dermatol Res 2020; DOI: 10.1007/s00403-020-02114-w. https://doi.org/10.1007/s00403-020-02114-w |
||||
| 16 Reinehr CPH, Bakos RM: Actinic keratoses: review of clinical, dermoscopic, and therapeutic aspects. An Bras Dermatol 2019;94:637-657. https://doi.org/10.1016/j.abd.2019.10.004 |
||||
| 17 Surjana D, Halliday GM, Martin AJ, Moloney FJ, Damian DL: Oral nicotinamide reduces actinic keratoses in phase II double-blinded randomized controlled trials. J Invest Dermatol 2012;132:1497-1500. https://doi.org/10.1038/jid.2011.459 |
||||
| 18 Chen AC, Martin AJ, Choy B, Fernandez-Penas P, Dalziell RA, McKenzie CA, Scolyer RA, Dhillon HM, Vardy JL, Kricker A, St George G, Chinniah N, Halliday GM, Damian DL: A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N Engl J Med 2015;373:1618-1626. https://doi.org/10.1056/NEJMoa1506197 |
||||
| 19 Krynitz B, Edgren G, Lindelof B, Baecklund E, Brattstom C, Wilczek H, Smedby KE: Risk of skin cancer and other malignancies in kidney, liver, heart and lung transplant recipients 1970 to 2008-A Swedish population-based study. Int J Cancer 2012:1429-1438. https://doi.org/10.1002/ijc.27765 |
||||
| 20 Jiyad Z, Olsen CM, Burke MT, Isbel NM, Green AC: Azathioprine and Risk of Skin Cancer in Organ Transplant Recipients: Systematic Review and Meta-Analysis. Am J Transplant 2016;16:3490-3503. https://doi.org/10.1111/ajt.13863 |
||||
| 21 Ume AC, Pugh JM, Kemp MG, Williams CR: Calcineurin inhibitor (CNI)-associated skin cancers: New insights on exploring mechanisms by which CNIs downregulate DNA repair machinery. Photodermatol Photoimmunol Photomed 2020;36:433-440. https://doi.org/10.1111/phpp.12600 |
||||
| 22 Murray SL, Daly FE, O'Kelly P, O'Leary E, Deady S, O'Neill JP, Dudley A, Rutledge NR, McCormick A, Houlihan DD, Williams Y, Morris PG, Ni Raghallaigh S, Moloney FJ, Sexton DJ, Conlon PJ: The impact of switching to mTOR inhibitor-based immunosuppression on long-term non-melanoma skin cancer incidence and renal function in kidney and liver transplant recipients. Ren Fail 2020;42:607-612. https://doi.org/10.1080/0886022X.2020.1785499 |
||||
| 23 Geissler EK: Skin cancer in solid organ transplant recipients: are mTOR inhibitors a game changer? Transplant Res 2015;4:1. https://doi.org/10.1186/s13737-014-0022-4 |
||||
| 24 Willenbrink TJ, Jambusaria-Pahlajani A, Arron S, Seckin D, Harwood CA, Proby CM: Treatment approaches in immunosuppressed patients with advanced cutaneous squamous cell carcinoma. J Eur Acad Dermatol Venereol 2019;33:57-60. https://doi.org/10.1111/jdv.15843 |
||||
| 25 Schauder DM, Kim J, Nijhawan RI: Evaluation of the Use of Capecitabine for the Treatment and Prevention of Actinic Keratoses, Squamous Cell Carcinoma, and Basal Cell Carcinoma: A Systematic Review. JAMA Dermatol 2020;156:1117-1124. https://doi.org/10.1001/jamadermatol.2020.2327 |
||||
| 26 van Lee CB, Roorda BM, Wakkee M, Voorham Q, Mooyaart AL, de Vijlder HC, Nijsten T, van den Bos RR: Recurrence rates of cutaneous squamous cell carcinoma of the head and neck after Mohs micrographic surgery vs. standard excision: a retrospective cohort study. Br J Dermatol 2019;181:338-343. https://doi.org/10.1111/bjd.17188 |
||||
| 27 Potenza C, Bernardini N, Balduzzi V, Losco L, Mambrin A, Marchesiello A, Tolino E, Zuber S, Skroza N, Proietti I: A Review of the Literature of Surgical and Nonsurgical Treatments of Invasive Squamous Cells Carcinoma. Biomed Res Int 2018;2018:9489163. https://doi.org/10.1155/2018/9489163 |
||||
| 28 Schmults CD, Karia PS, Carter JB, Han J, Qureshi AA: Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: a 10-year, single-institution cohort study. JAMA Dermatol 2013;149:541-547. https://doi.org/10.1001/jamadermatol.2013.2139 |
||||
| 29 Brantsch KD, Meisner C, Schonfisch B, Trilling B, Wehner-Caroli J, Rocken M, Breuninger H: Analysis of risk factors determining prognosis of cutaneous squamous-cell carcinoma: a prospective study. Lancet Oncol 2008;9:713-720. https://doi.org/10.1016/S1470-2045(08)70178-5 |
||||
| 30 Brinkman JN, Hajder E, van der Holt B, Den Bakker MA, Hovius SE, Mureau MA: The Effect of Differentiation Grade of Cutaneous Squamous Cell Carcinoma on Excision Margins, Local Recurrence, Metastasis, and Patient Survival: A Retrospective Follow-Up Study. Ann Plast Surg 2015;75:323-326. https://doi.org/10.1097/SAP.0000000000000110 |
||||
| 31 Thompson AK, Kelley BF, Prokop LJ, Murad MH, Baum CL: Risk Factors for Cutaneous Squamous Cell Carcinoma Recurrence, Metastasis, and Disease-Specific Death: A Systematic Review and Meta-analysis. JAMA Dermatol 2016;152:419-428. https://doi.org/10.1001/jamadermatol.2015.4994 |
||||
| 32 Singh B, Carpenter G, Coffey RJ: EGF receptor ligands: recent advances. F1000Res 2016; DOI: 10.12688/f1000research.9025.1. https://doi.org/10.12688/f1000research.9025.1 |
||||
| 33 Xu Y, Shao Y, Zhou J, Voorhees JJ, Fisher GJ: Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J Cell Biochem 2009;107:873-880. https://doi.org/10.1002/jcb.22195 |
||||
| 34 El-Abaseri TB, Fuhrman J, Trempus C, Shendrik I, Tennant RW, Hansen LA: Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res 2005;65:3958-3965. https://doi.org/10.1158/0008-5472.CAN-04-2204 |
||||
| 35 El-Abaseri TB, Putta S, Hansen LA: Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis 2006;27:225-231. https://doi.org/10.1093/carcin/bgi220 |
||||
| 36 El-Abaseri TB, Hammiller B, Repertinger SK, Hansen LA: The epidermal growth factor receptor increases cytokine production and cutaneous inflammation in response to ultraviolet irradiation. ISRN Dermatol 2013;2013:848705. https://doi.org/10.1155/2013/848705 |
||||
| 37 Oi N, Chen H, Ok Kim M, Lubet RA, Bode AM, Dong Z: Taxifolin suppresses UV-induced skin carcinogenesis by targeting EGFR and PI3K. Cancer Prev Res (Phila) 2012;5:1103-1114. https://doi.org/10.1158/1940-6207.CAPR-11-0397 |
||||
| 38 Toll A, Salgado R, Yebenes M, Martin-Ezquerra G, Gilaberte M, Baro T, Sole F, Alameda F, Espinet B, Pujol RM: Epidermal growth factor receptor gene numerical aberrations are frequent events in actinic keratoses and invasive cutaneous squamous cell carcinomas. Exp Dermatol 2010;19:151-153. https://doi.org/10.1111/j.1600-0625.2009.01028.x |
||||
| 39 Bito T, Sumita N, Ashida M, Budiyanto A, Ueda M, Ichihashi M, Tokura Y, Nishigori C: Inhibition of Epidermal Growth Factor Receptor and PI3K/Akt Signaling Suppresses Cell Proliferation and Survival through Regulation of Stat3 Activation in Human Cutaneous Squamous Cell Carcinoma. J Skin Cancer 2011;2011:874571. https://doi.org/10.1155/2011/874571 |
||||
| 40 Canueto J, Cardenoso E, Garcia JL, Santos-Briz A, Castellanos-Martin A, Fernandez-Lopez E, Blanco Gomez A, Perez-Losada J, Roman-Curto C: Epidermal growth factor receptor expression is associated with poor outcome in cutaneous squamous cell carcinoma. Br J Dermatol 2017;176:1279-1287. https://doi.org/10.1111/bjd.14936 |
||||
| 41 Kitamura S, Maeda T, Yanagi T: Vandetanib inhibits cell growth in EGFR-expressing cutaneous squamous cell carcinoma. Biochem Biophys Res Commun 2020;531:396-401. https://doi.org/10.1016/j.bbrc.2020.07.111 |
||||
| 42 Ch'ng S, Low I, Ng D, Brasch H, Sullivan M, Davis P, Tan ST: Epidermal growth factor receptor: a novel biomarker for aggressive head and neck cutaneous squamous cell carcinoma. Hum Pathol 2008;39:344-349. https://doi.org/10.1016/j.humpath.2007.07.004 |
||||
| 43 Egashira S, Jinnin M, Ajino M, Shimozono N, Okamoto S, Tasaki Y, Hirano A, Ide M, Kajihara I, Aoi J, Harada M, Igata T, Masuguchi S, Fukushima S, Ihn H: Chronic sun exposure-related fusion oncogenes EGFR-PPARGC1A in cutaneous squamous cell carcinoma. Sci Rep 2017;7:12654. https://doi.org/10.1038/s41598-017-12836-z |
||||
| 44 Al-Rohil RN, Tarasen AJ, Carlson JA, Wang K, Johnson A, Yelensky R, Lipson D, Elvin JA, Vergilio JA, Ali SM, Suh J, Miller VA, Stephens PJ, Ganesan P, Janku F, Karp DD, Subbiah V, Mihm MC, Ross JS: Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016;122:249-257. https://doi.org/10.1002/cncr.29738 |
||||
| 45 Montaudie H, Viotti J, Combemale P, Dutriaux C, Dupin N, Robert C, Mortier L, Kaphan R, Duval-Modeste AB, Dalle S, De Quatrebarbes J, Stefan A, Brunet-Possenti F, Kogay M, Picard-Gauci A, Poissonnet G, Peyrade F: Cetuximab is efficient and safe in patients with advanced cutaneous squamous cell carcinoma: a retrospective, multicentre study. Oncotarget 2020;11:378-385. https://doi.org/10.18632/oncotarget.27434 |
||||
| 46 Joseph K, Alkaabi K, Warkentin H, Ghosh S, Jha N, Smylie M, Walker J: Cetuximab-radiotherapy combination in the management of locally advanced cutaneous squamous cell carcinoma. J Med Imaging Radiat Oncol 2019;63:257-263. https://doi.org/10.1111/1754-9485.12842 |
||||
| 47 Trodello C, Higgins S, Ahadiat O, Ragab O, In G, Hawkins M, Wysong A: Cetuximab as a Component of Multimodality Treatment of High-Risk Cutaneous Squamous Cell Carcinoma: A Retrospective Analysis From a Single Tertiary Academic Medical Center. Dermatol Surg 2019;45:254-267. https://doi.org/10.1097/DSS.0000000000001755 |
||||
| 48 Jenni D, Karpova MB, Muhleisen B, Mangana J, Dreier J, Hafner J, Dummer R: A prospective clinical trial to assess lapatinib effects on cutaneous squamous cell carcinoma and actinic keratosis. ESMO Open 2016;1:e000003. https://doi.org/10.1136/esmoopen-2015-000003 |
||||
| 49 William WN, Jr., Feng L, Ferrarotto R, Ginsberg L, Kies M, Lippman S, Glisson B, Kim ES: Gefitinib for patients with incurable cutaneous squamous cell carcinoma: A single-arm phase II clinical trial. J Am Acad Dermatol 2017;77:1110-1113 e1112. https://doi.org/10.1016/j.jaad.2017.07.048 |
||||
| 50 Gold KA, Kies MS, William WN, Jr., Johnson FM, Lee JJ, Glisson BS: Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: A single-arm phase 2 clinical trial. Cancer 2018;124:2169-2173. https://doi.org/10.1002/cncr.31346 |
||||
| 51 McHugh A, Fernandes K, Chinner N, Ibrahim AFM, Garg AK, Boag G, Hepburn LA, Proby CM, Leigh IM, Saville MK: The Identification of Potential Therapeutic Targets for Cutaneous Squamous Cell Carcinoma. J Invest Dermatol 2020;140:1154-1165 e1155. https://doi.org/10.1016/j.jid.2019.09.024 |
||||
| 52 Ise K, Nakamura K, Nakao K, Shimizu S, Harada H, Ichise T, Miyoshi J, Gondo Y, Ishikawa T, Aiba A, Katsuki M: Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene 2000;19:2951-2956. https://doi.org/10.1038/sj.onc.1203600 |
||||
| 53 Drosten M, Lechuga CG, Barbacid M: Ras signaling is essential for skin development. Oncogene 2014;33:2857-2865. https://doi.org/10.1038/onc.2013.254 |
||||
| 54 Campos MA, Macedo S, Fernandes MS, Pestana A, Pardal J, Batista R, Vinagre J, Sanches A, Baptista A, Lopes JM, Soares P: Prognostic Significance of RAS Mutations and P53 Expression in Cutaneous Squamous Cell Carcinomas. Genes (Basel) 2020;11:751. https://doi.org/10.3390/genes11070751 |
||||
| 55 Lee CS, Bhaduri A, Mah A, Johnson WL, Ungewickell A, Aros CJ, Nguyen CB, Rios EJ, Siprashvili Z, Straight A, Kim J, Aasi SZ, Khavari PA: Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat Genet 2014;46:1060-1062. https://doi.org/10.1038/ng.3091 |
||||
| 56 Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina TV, Enikolopov G, Muthuswamy SK, Mills AA: DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011;8:164-176. https://doi.org/10.1016/j.stem.2010.12.009 |
||||
| 57 Abel EL, Angel JM, Kiguchi K, DiGiovanni J: Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 2009;4:1350-1362. https://doi.org/10.1038/nprot.2009.120 |
||||
| 58 Schwarz M, Munzel PA, Braeuning A: Non-melanoma skin cancer in mouse and man. Arch Toxicol 2013;87:783-798. https://doi.org/10.1007/s00204-012-0998-9 |
||||
| 59 Wong CE, Yu JS, Quigley DA, To MD, Jen KY, Huang PY, Del Rosario R, Balmain A: Inflammation and Hras signaling control epithelial-mesenchymal transition during skin tumor progression. Genes Dev 2013;27:670-682. https://doi.org/10.1101/gad.210427.112 |
||||
| 60 Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, Fang M, Tao S, Green CL, Khavari PA: CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med 2002;8:1105-1114. https://doi.org/10.1038/nm779 |
||||
| 61 Golomb L, Sagiv A, Pateras IS, Maly A, Krizhanovsky V, Gorgoulis VG, Oren M, Ben-Yehuda A: Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ 2015;22:1764-1774. https://doi.org/10.1038/cdd.2015.21 |
||||
| 62 Liceras-Boillos P, Jimeno D, Garcia-Navas R, Lorenzo-Martin LF, Menacho-Marquez M, Segrelles C, Gomez C, Calzada N, Fuentes-Mateos R, Paramio JM, Bustelo XR, Baltanas FC, Santos E: Differential Role of the RasGEFs Sos1 and Sos2 in Mouse Skin Homeostasis and Carcinogenesis. Mol Cell Biol 2018;38:e00049-18. https://doi.org/10.1128/MCB.00049-18 |
||||
| 63 Fonseca LL, Yang WS, Geerts D, Turkson J, Ji J, Ramos JW: RasGRP1 induces autophagy and transformation-associated changes in primary human keratinocytes. Transl Oncol 2021;14:100880. https://doi.org/10.1016/j.tranon.2020.100880 |
||||
| 64 Maurer G, Tarkowski B, Baccarini M: Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011;30:3477-3488. https://doi.org/10.1038/onc.2011.160 |
||||
| 65 Menzies AM, Kefford RF, Long GV: Paradoxical oncogenesis: are all BRAF inhibitors equal? Pigment Cell Melanoma Res 2013;26:611-615. https://doi.org/10.1111/pcmr.12132 |
||||
| 66 Anforth R, Menzies A, Byth K, Carlos G, Chou S, Sharma R, Scolyer RA, Kefford R, Long GV, Fernandez-Penas P: Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J Am Acad Dermatol 2015;72:809-815.e1. https://doi.org/10.1016/j.jaad.2015.01.018 |
||||
| 67 Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, Martin M, Yang H, Wang Q, Hilton H, Hang JS, Noe J, Lambros M, et al.: RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207-215. https://doi.org/10.1056/NEJMoa1105358 |
||||
| 68 Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R: Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010;140:209-221. https://doi.org/10.1016/j.cell.2009.12.040 |
||||
| 69 Scholl FA, Dumesic PA, Barragan DI, Harada K, Bissonauth V, Charron J, Khavari PA: Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Dev Cell 2007;12:615-629. https://doi.org/10.1016/j.devcel.2007.03.009 |
||||
| 70 Scholl FA, Dumesic PA, Khavari PA: Mek1 alters epidermal growth and differentiation. Cancer Res 2004;64:6035-6040. https://doi.org/10.1158/0008-5472.CAN-04-0017 |
||||
| 71 Li YY, Hanna GJ, Laga AC, Haddad RI, Lorch JH, Hammerman PS: Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin Cancer Res 2015;21:1447-1456. https://doi.org/10.1158/1078-0432.CCR-14-1773 |
||||
| 72 Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, Tsai KY, Curry JL, Tetzlaff MT, Lai SY, Yu J, Muzny DM, Doddapaneni H, Shinbrot E, Covington KR, Zhang J, Seth S, Caulin C, Clayman GL, El-Naggar AK, et al.: Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res 2014;20:6582-6592. https://doi.org/10.1158/1078-0432.CCR-14-1768 |
||||
| 73 Demehri S, Turkoz A, Kopan R: Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009;16:55-66. https://doi.org/10.1016/j.ccr.2009.05.016 |
||||
| 74 Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F: Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 2003;33:416-421. https://doi.org/10.1038/ng1099 |
||||
| 75 Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, Millar SE, Pear WS, Parmacek MS: Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res 2006;66:7438-7444. https://doi.org/10.1158/0008-5472.CAN-06-0793 |
||||
| 76 Pan L, Lemieux ME, Thomas T, Rogers JM, Lipper CH, Lee W, Johnson C, Sholl LM, South AP, Marto JA, Adelmant GO, Blacklow SC, Aster JC: IER5, a DNA damage response gene, is required for Notch-mediated induction of squamous cell differentiation. Elife 2020;9:e58081. https://doi.org/10.7554/eLife.58081 |
||||
| 77 Quan XX, Hawk NV, Chen W, Coupar J, Lee SK, Petersen DW, Meltzer PS, Montemarano A, Braun M, Chen Z, Van Waes C: Targeting Notch1 and IKKalpha Enhanced NF-kappaB Activation in CD133(+) Skin Cancer Stem Cells. Mol Cancer Ther 2018;17:2034-2048. https://doi.org/10.1158/1535-7163.MCT-17-0421 |
||||
| 78 Wu F, Weigel KJ, Zhou H, Wang XJ: Paradoxical roles of TGF-beta signaling in suppressing and promoting squamous cell carcinoma. Acta Biochim Biophys Sin (Shanghai) 2018;50:98-105. https://doi.org/10.1093/abbs/gmx127 |
||||
| 79 Oshimori N, Oristian D, Fuchs E: TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015;160:963-976. https://doi.org/10.1016/j.cell.2015.01.043 |
||||
| 80 Cammareri P, Rose AM, Vincent DF, Wang J, Nagano A, Libertini S, Ridgway RA, Athineos D, Coates PJ, McHugh A, Pourreyron C, Dayal JH, Larsson J, Weidlich S, Spender LC, Sapkota GP, Purdie KJ, Proby CM, Harwood CA, Leigh IM, et al.: Inactivation of TGFbeta receptors in stem cells drives cutaneous squamous cell carcinoma. Nat Commun 2016;7:12493. https://doi.org/10.1038/ncomms12493 |
||||
| 81 Rose AM, Spender LC, Stephen C, Mitchell A, Rickaby W, Bray S, Evans AT, Dayal J, Purdie KJ, Harwood CA, Proby CM, Leigh IM, Coates PJ, Inman GJ: Reduced SMAD2/3 activation independently predicts increased depth of human cutaneous squamous cell carcinoma. Oncotarget 2018;9:14552-14566. https://doi.org/10.18632/oncotarget.24545 |
||||
| 82 Yang L, Mao C, Teng Y, Li W, Zhang J, Cheng X, Li X, Han X, Xia Z, Deng H, Yang X: Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer Res 2005;65:8671-8678. https://doi.org/10.1158/0008-5472.CAN-05-0800 |
||||
| 83 Qiao W, Li AG, Owens P, Xu X, Wang XJ, Deng CX: Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene 2006;25:207-217. https://doi.org/10.1038/sj.onc.1209029 |
||||
| 84 Hoot KE, Lighthall J, Han G, Lu SL, Li A, Ju W, Kulesz-Martin M, Bottinger E, Wang XJ: Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J Clin Invest 2008;118:2722-2732. https://doi.org/10.1172/JCI33713 |
||||
| 85 Li AG, Lu SL, Zhang MX, Deng C, Wang XJ: Smad3 knockout mice exhibit a resistance to skin chemical carcinogenesis. Cancer Res 2004;64:7836-7845. https://doi.org/10.1158/0008-5472.CAN-04-1331 |
||||
| 86 Li F, Bian L, Iriyama S, Jian Z, Fan B, Luo J, Wang DD, Young CD, Han G, Wang XJ: Smad7 Ameliorates TGF-beta-Mediated Skin Inflammation and Associated Wound Healing Defects but Not Susceptibility to Experimental Skin Carcinogenesis. J Invest Dermatol 2019;139:940-950. https://doi.org/10.1016/j.jid.2018.10.031 |
||||
| 87 Aschauer L, Muller PA: Novel targets and interaction partners of mutant p53 Gain-Of-Function. Biochem Soc Trans 2016;44:460-466. | ||||
| 88 Javor S, Gasparini G, Biatta CM, Cozzani E, Cabiddu F, Ravetti JL, Vellone VG, Parodi A: P53 staining index and zonal staining patterns in actinic keratoses. Arch Dermatol Res 2021;313:275-279. https://doi.org/10.1007/s00403-020-02104-y |
||||
| 89 Bakshi A, Shafi R, Nelson J, Cantrell WC, Subhadarshani S, Andea A, Athar M, Elmets CA: The clinical course of actinic keratosis correlates with underlying molecular mechanisms. Br J Dermatol 2020;182:995-1002. https://doi.org/10.1111/bjd.18338 |
||||
| 90 Marinescu A, Stepan AE, Margaritescu C, Marinescu AM, Zavoi RE, Simionescu CE, Niculescu M: P53, p16 and Ki67 immunoexpression in cutaneous squamous cell carcinoma and its precursor lesions. Rom J Morphol Embryol 2016;57:691-696. | ||||
| 91 Kusters-Vandevelde HV, Van Leeuwen A, Verdijk MA, de Koning MN, Quint WG, Melchers WJ, Ligtenberg MJ, Blokx WA: CDKN2A but not TP53 mutations nor HPV presence predict poor outcome in metastatic squamous cell carcinoma of the skin. Int J Cancer 2010;126:2123-2132. https://doi.org/10.1002/ijc.24871 |
||||
| 92 Nguyen D, Liao W, Zeng SX, Lu H: Reviving the guardian of the genome: Small molecule activators of p53. Pharmacol Ther 2017;178:92-108. https://doi.org/10.1016/j.pharmthera.2017.03.013 |
||||
| 93 Duffy MJ, Synnott NC, O'Grady S, Crown J: Targeting p53 for the treatment of cancer. Semin Cancer Biol 2020; DOI: 10.1016/j.semcancer.2020.07.005. https://doi.org/10.1016/j.semcancer.2020.07.005 |
||||
| 94 Pillai RN, Fennell DA, Kovcin V, Ciuleanu TE, Ramlau R, Kowalski D, Schenker M, Yalcin I, Teofilovici F, Vukovic VM, Ramalingam SS: Randomized Phase III Study of Ganetespib, a Heat Shock Protein 90 Inhibitor, With Docetaxel Versus Docetaxel in Advanced Non-Small-Cell Lung Cancer (GALAXY-2). J Clin Oncol 2020;38:613-622. https://doi.org/10.1200/JCO.19.00816 |
||||
| 95 Soares E, Zhou H: Master regulatory role of p63 in epidermal development and disease. Cell Mol Life Sci 2018;75:1179-1190. https://doi.org/10.1007/s00018-017-2701-z |
||||
| 96 Truong AB, Khavari PA: Control of keratinocyte proliferation and differentiation by p63. Cell cycle (Georgetown, Tex) 2007;6:295-299. https://doi.org/10.4161/cc.6.3.3753 |
||||
| 97 Wrone DA, Yoo S, Chipps LK, Moy RL: The expression of p63 in actinic keratoses, seborrheic keratosis, and cutaneous squamous cell carcinomas. Dermatol Surg 2004;30:1299-1302. https://doi.org/10.1111/j.1524-4725.2004.30403.x |
||||
| 98 Liefer KM, Koster MI, Wang XJ, Yang A, McKeon F, Roop DR: Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res 2000;60:4016-4020. | ||||
| 99 Devos M, Gilbert B, Denecker G, Leurs K, Mc Guire C, Lemeire K, Hochepied T, Vuylsteke M, Lambert J, Van Den Broecke C, Libbrecht L, Haigh J, Berx G, Lippens S, Vandenabeele P, Declercq W: Elevated DeltaNp63alpha Levels Facilitate Epidermal and Biliary Oncogenic Transformation. J Invest Dermatol 2017;137:494-505. https://doi.org/10.1016/j.jid.2016.09.026 |
||||
| 100 Rokudai S, Li Y, Otaka Y, Fujieda M, Owens DM, Christiano AM, Nishiyama M, Prives C: STXBP4 regulates APC/C-mediated p63 turnover and drives squamous cell carcinogenesis. Proc Natl Acad Sci U S A 2018;115:E4806-E4814. https://doi.org/10.1073/pnas.1718546115 |
||||
| 101 Choo MK, Kraft S, Missero C, Park JM: The protein kinase p38alpha destabilizes p63 to limit epidermal stem cell frequency and tumorigenic potential. Sci Signal 2018;11:eaau0727. https://doi.org/10.1126/scisignal.aau0727 |
||||
| 102 Wang L, Xia W, Chen H, Xiao ZX: DeltaNp63alpha modulates phosphorylation of p38 MAP kinase in regulation of cell cycle progression and cell growth. Biochem Biophys Res Commun 2019;509:784-789. https://doi.org/10.1016/j.bbrc.2018.12.185 |
||||
| 103 Ha L, Ponnamperuma RM, Jay S, Ricci MS, Weinberg WC: Dysregulated DeltaNp63alpha inhibits expression of Ink4a/arf, blocks senescence, and promotes malignant conversion of keratinocytes. PLoS One 2011;6:e21877. https://doi.org/10.1371/journal.pone.0021877 |
||||
| 104 Maurelli R, Zambruno G, Guerra L, Abbruzzese C, Dimri G, Gellini M, Bondanza S, Dellambra E: Inactivation of p16INK4a (inhibitor of cyclin-dependent kinase 4A) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J 2006;20:1516-1518. https://doi.org/10.1096/fj.05-4480fje |
||||
| 105 Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, Biernaskie JA, Sinha S, Prives C, Pevny LH, Miller FD, Flores ER: TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 2009;5:64-75. https://doi.org/10.1016/j.stem.2009.04.003 |
||||
| 106 Park E, Kim H, Kim JM, Primack B, Vidal-Cardenas S, Xu Y, Price BD, Mills AA, D'Andrea AD: FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol Cell 2013;50:908-918. https://doi.org/10.1016/j.molcel.2013.05.017 |
||||
| 107 Davis AJ, Tsinkevich M, Rodencal J, Abbas HA, Su XH, Gi YJ, Fang B, Rajapakshe K, Coarfa C, Gunaratne PH, Koomen JM, Tsai KY, Flores ER: TAp63-Regulated miRNAs Suppress Cutaneous Squamous Cell Carcinoma through Inhibition of a Network of Cell-Cycle Genes. Cancer Res 2020;80:2484-2497. https://doi.org/10.1158/0008-5472.CAN-19-1892 |
||||
| 108 Beeler JS, Marshall CB, Gonzalez-Ericsson PI, Shaver TM, Santos Guasch GL, Lea ST, Johnson KN, Jin H, Venters BJ, Sanders ME, Pietenpol JA: p73 regulates epidermal wound healing and induced keratinocyte programming. PLoS One 2019;14:e0218458. https://doi.org/10.1371/journal.pone.0218458 |
||||
| 109 Botchkarev VA: Epigenetic Regulation of Epidermal Development and Keratinocyte Differentiation. J Investig Dermatol Symp Proc 2015;17:18-19. https://doi.org/10.1038/jidsymp.2015.15 |
||||
| 110 Johnson J, Lagowski J, Sundberg A, Lawson S, Liu Y, Kulesz-Martin M: p73 loss triggers conversion to squamous cell carcinoma reversible upon reconstitution with TAp73alpha. Cancer Res 2007;67:7723-7730. https://doi.org/10.1158/0008-5472.CAN-07-1195 |
||||
| 111 Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F, Jacks T: Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005;7:363-373. https://doi.org/10.1016/j.ccr.2005.02.019 |
||||
| 112 Shen Y, Xu J, Jin J, Tang H, Liang J: Cyclin D1 expression in Bowen's disease and cutaneous squamous cell carcinoma. Mol Clin Oncol 2014;2:545-548. https://doi.org/10.3892/mco.2014.273 |
||||
| 113 Liang SB, Furihata M, Takeuchi T, Iwata J, Chen BK, Sonobe H, Ohtsuki Y: Overexpression of cyclin D1 in nonmelanocytic skin cancer. Virchows Arch 2000;436:370-376. https://doi.org/10.1007/s004280050461 |
||||
| 114 Wu J, Lu WY, Cui LL: Clinical significance of STAT3 and MAPK phosphorylation, and the protein expression of cyclin D1 in skin squamous cell carcinoma tissues. Mol Med Rep 2015;12:8129-8134. https://doi.org/10.3892/mmr.2015.4460 |
||||
| 115 Huang K, Huang C, Shan K, Chen J, Li H: Significance of PC cell-derived growth factor and cyclin D1 expression in cutaneous squamous cell carcinoma. Clin Exp Dermatol 2012;37:411-417. https://doi.org/10.1111/j.1365-2230.2011.04275.x |
||||
| 116 Utikal J, Udart M, Leiter U, Kaskel P, Peter RU, Krahn G: Numerical abnormalities of the Cyclin D1 gene locus on chromosome 11q13 in non-melanoma skin cancer. Cancer Lett 2005;219:197-204. https://doi.org/10.1016/j.canlet.2004.07.011 |
||||
| 117 Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, Tennant RW, Weinberg RA, Yuspa SH, Conti CJ: Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev 1998;12:2469-2474. https://doi.org/10.1101/gad.12.16.2469 |
||||
| 118 Yamamoto H, Ochiya T, Takeshita F, Toriyama-Baba H, Hirai K, Sasaki H, Sasaki H, Sakamoto H, Yoshida T, Saito I, Terada M: Enhanced skin carcinogenesis in cyclin D1-conditional transgenic mice: cyclin D1 alters keratinocyte response to calcium-induced terminal differentiation. Cancer Res 2002;62:1641-1647. | ||||
| 119 Zhou M, Zhou L, Zheng L, Guo L, Wang Y, Liu H, Ou C, Ding Z: miR-365 promotes cutaneous squamous cell carcinoma (CSCC) through targeting nuclear factor I/B (NFIB). PLoS One 2014;9:e100620. https://doi.org/10.1371/journal.pone.0100620 |
||||
| 120 Zhou L, Wang Y, Ou C, Lin Z, Wang J, Liu H, Zhou M, Ding Z: microRNA-365-targeted nuclear factor I/B transcriptionally represses cyclin-dependent kinase 6 and 4 to inhibit the progression of cutaneous squamous cell carcinoma. Int J Biochem Cell Biol 2015;65:182-191. https://doi.org/10.1016/j.biocel.2015.06.009 |
||||
| 121 Shi X, Li H, Shi A, Yao H, Ke K, Dong C, Zhu Y, Qin Y, Ding Y, He YH, Liu X, Li L, Lei L, Hai Q, Chen W, Leung KS, Wong MH, Kung HF, Lin MC: Discovery of rafoxanide as a dual CDK4/6 inhibitor for the treatment of skin cancer. Oncol Rep 2018;40:1592-1600. https://doi.org/10.3892/or.2018.6533 |
||||
| 122 Lazo de la Vega L, Bick N, Hu K, Rahrig SE, Silva CD, Matayoshi S, Picciarelli P, Wang X, Sugar A, Soong HK, Mian SI, Robinson DR, Chinnaiyan AM, Demirci H, Daniels AB, Worden F, Eberhart CG, Tomlins SA, Rao RC, Harms PW: Invasive squamous cell carcinomas and precursor lesions on UV-exposed epithelia demonstrate concordant genomic complexity in driver genes. Mod Pathol 2020;33:2280-2294. https://doi.org/10.1038/s41379-020-0571-7 |
||||
| 123 Costa C, Santos M, Martinez-Fernandez M, Duenas M, Lorz C, Garcia-Escudero R, Paramio JM: E2F1 loss induces spontaneous tumour development in Rb-deficient epidermis. Oncogene 2013;32:2937-2951. https://doi.org/10.1038/onc.2012.316 |
||||
| 124 Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP, Proby CM: p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Invest Dermatol 2004;122:1284-1292. https://doi.org/10.1111/j.0022-202X.2004.22501.x |
||||
| 125 Ahmad I, Guroji P, DeBrot AH, Manapragada PP, Katiyar SK, Elmets CA, Yusuf N: Loss of INK4a/Arf gene enhances ultraviolet radiation-induced cutaneous tumor development. Exp Dermatol 2017;26:1018-1025. https://doi.org/10.1111/exd.13356 |
||||
| 126 Zhao R, Choi BY, Lee MH, Bode AM, Dong Z: Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016;8:30-39. https://doi.org/10.1016/j.ebiom.2016.04.017 |
||||
| 127 Zanet J, Pibre S, Jacquet C, Ramirez A, de Alboran IM, Gandarillas A: Endogenous Myc controls mammalian epidermal cell size, hyperproliferation, endoreplication and stem cell amplification. J Cell Sci 2005;118:1693-1704. https://doi.org/10.1242/jcs.02298 |
||||
| 128 Oskarsson T, Essers MA, Dubois N, Offner S, Dubey C, Roger C, Metzger D, Chambon P, Hummler E, Beard P, Trumpp A: Skin epidermis lacking the c-Myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21Cip1 gene. Genes Dev 2006;20:2024-2029. https://doi.org/10.1101/gad.381206 |
||||
| 129 Pelengaris S, Littlewood T, Khan M, Elia G, Evan G: Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell 1999;3:565-577. https://doi.org/10.1016/S1097-2765(00)80350-0 |
||||
| 130 Toll A, Salgado R, Yebenes M, Martin-Ezquerra G, Gilaberte M, Baro T, Sole F, Alameda F, Espinet B, Pujol RM: MYC gene numerical aberrations in actinic keratosis and cutaneous squamous cell carcinoma. Br J Dermatol 2009;161:1112-1118. https://doi.org/10.1111/j.1365-2133.2009.09351.x |
||||
| 131 Lohcharoenkal W, Harada M, Loven J, Meisgen F, Landen NX, Zhang L, Lapins J, Mahapatra KD, Shi H, Nissinen L, Kahari VM, Stahle M, Sonkoly E, Grander D, Arsenian-Henriksson M, Pivarcsi A: MicroRNA-203 Inversely Correlates with Differentiation Grade, Targets c-MYC, and Functions as a Tumor Suppressor in cSCC. J Invest Dermatol 2016;136:2485-2494. https://doi.org/10.1016/j.jid.2016.06.630 |
||||
| 132 Wang X, Langer EM, Daniel CJ, Janghorban M, Wu V, Wang XJ, Sears RC: Altering MYC phosphorylation in the epidermis increases the stem cell population and contributes to the development, progression, and metastasis of squamous cell carcinoma. Oncogenesis 2020;9:79. https://doi.org/10.1038/s41389-020-00261-3 |
||||
| 133 Audia JE, Campbell RM: Histone Modifications and Cancer. Cold Spring Harb Perspect Biol 2016;8:a019521. https://doi.org/10.1101/cshperspect.a019521 |
||||
| 134 Vandiver AR, Irizarry RA, Hansen KD, Garza LA, Runarsson A, Li X, Chien AL, Wang TS, Leung SG, Kang S, Feinberg AP: Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol 2015;16:80. https://doi.org/10.1186/s13059-015-0644-y |
||||
| 135 Li L, Li F, Xia Y, Yang X, Lv Q, Fang F, Wang Q, Bu W, Wang Y, Zhang K, Wu Y, Shen J, Jiang M: UVB induces cutaneous squamous cell carcinoma progression by de novo ID4 methylation via methylation regulating enzymes. EBioMedicine 2020;57:102835. https://doi.org/10.1016/j.ebiom.2020.102835 |
||||
| 136 Hervas-Marin D, Higgins F, Sanmartin O, Lopez-Guerrero JA, Bano MC, Igual JC, Quilis I, Sandoval J: Genome wide DNA methylation profiling identifies specific epigenetic features in high-risk cutaneous squamous cell carcinoma. PLoS One 2019;14:e0223341. https://doi.org/10.1371/journal.pone.0223341 |
||||
| 137 Rodriguez-Paredes M, Bormann F, Raddatz G, Gutekunst J, Lucena-Porcel C, Kohler F, Wurzer E, Schmidt K, Gallinat S, Wenck H, Rowert-Huber J, Denisova E, Feuerbach L, Park J, Brors B, Herpel E, Nindl I, Hofmann TG, Winnefeld M, Lyko F: Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat Commun 2018;9:577. https://doi.org/10.1038/s41467-018-03025-1 |
||||
| 138 Egolf S, Aubert Y, Doepner M, Anderson A, Maldonado-Lopez A, Pacella G, Lee J, Ko EK, Zou J, Lan Y, Simpson CL, Ridky T, Capell BC: LSD1 Inhibition Promotes Epithelial Differentiation through Derepression of Fate-Determining Transcription Factors. Cell Rep 2019;28:1981-1992 e1987. https://doi.org/10.1016/j.celrep.2019.07.058 |
||||
| 139 Hernandez-Ruiz E, Toll A, Garcia-Diez I, Andrades E, Ferrandiz-Pulido C, Masferrer E, Yebenes M, Jaka A, Gimeno J, Gimeno R, Garcia-Patos V, Pujol RM, Hernandez-Munoz I: The Polycomb proteins RING1B and EZH2 repress the tumoral pro-inflammatory function in metastasizing primary cutaneous squamous cell carcinoma. Carcinogenesis 2018;39:503-513. https://doi.org/10.1093/carcin/bgy016 |
||||
| 140 Avgustinova A, Symeonidi A, Castellanos A, Urdiroz-Urricelqui U, Sole-Boldo L, Martin M, Perez-Rodriguez I, Prats N, Lehner B, Supek F, Benitah SA: Loss of G9a preserves mutation patterns but increases chromatin accessibility, genomic instability and aggressiveness in skin tumours. Nat Cell Biol 2018;20:1400-1409. https://doi.org/10.1038/s41556-018-0233-x |
||||
| 141 Watt SA, Purdie KJ, den Breems NY, Dimon M, Tucker S, Arron ST, McHugh A, Xue DJ, Dayal JH, Harwood CA, Leigh IM, Proby CM, South AP: CREBBP mutation in human cutaneous squamous cell carcinoma. Exp Dermatol 2016;25:650-651. https://doi.org/10.1111/exd.13044 |
||||
| 142 Chen MK, Cai MY, Luo RZ, Tian X, Liao QM, Zhang XY, Han JD: Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. Br J Dermatol 2015;172:111-119. https://doi.org/10.1111/bjd.13226 |
||||
| 143 Bosic MM, Brasanac DC, Stojkovic-Filipovic JM, Zaletel IV, Gardner JM, Cirovic SL: Expression of p300 and p300/CBP associated factor (PCAF) in actinic keratosis and squamous cell carcinoma of the skin. Exp Mol Pathol 2016;100:378-385. https://doi.org/10.1016/j.yexmp.2016.03.006 |
||||
| 144 Ichise T, Yoshida N, Ichise H: CBP/p300 antagonises EGFR-Ras-Erk signalling and suppresses increased Ras-Erk signalling-induced tumour formation in mice. J Pathol 2019;249:39-51. https://doi.org/10.1002/path.5279 |
||||
| 145 Zhang L, Shan X, Chen Q, Xu D, Fan X, Yu M, Yan Q, Liu J: Downregulation of HDAC3 by ginsenoside Rg3 inhibits epithelial-mesenchymal transition of cutaneous squamous cell carcinoma through c-Jun acetylation. J Cell Physiol 2019;234:22207-22219. https://doi.org/10.1002/jcp.28788 |
||||
| 146 Kalin JH, Eroglu A, Liu H, Holtzclaw WD, Leigh I, Proby CM, Fahey JW, Cole PA, Dinkova-Kostova AT: Investigation into the use of histone deacetylase inhibitor MS-275 as a topical agent for the prevention and treatment of cutaneous squamous cell carcinoma in an SKH-1 hairless mouse model. PLoS One 2019;14:e0213095. https://doi.org/10.1371/journal.pone.0213095 |
||||
| 147 Kurundkar D, Srivastava RK, Chaudhary SC, Ballestas ME, Kopelovich L, Elmets CA, Athar M: Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol Appl Pharmacol 2013;266:233-244. https://doi.org/10.1016/j.taap.2012.11.002 |
||||
| 148 Alfert A, Moreno N, Kerl K: The BAF complex in development and disease. Epigenetics Chromatin 2019;12:19. https://doi.org/10.1186/s13072-019-0264-y |
||||
| 149 Bock VL, Lyons JG, Huang XX, Jones AM, McDonald LA, Scolyer RA, Moloney FJ, Barnetson RS, Halliday GM: BRM and BRG1 subunits of the SWI/SNF chromatin remodelling complex are downregulated upon progression of benign skin lesions into invasive tumours. Br J Dermatol 2011;164:1221-1227. https://doi.org/10.1111/j.1365-2133.2011.10267.x |
||||
| 150 Hassan NM, Painter N, Howlett CR, Farrell AW, Di Girolamo N, Lyons JG, Halliday GM: Brm inhibits the proliferative response of keratinocytes and corneal epithelial cells to ultraviolet radiation-induced damage. PLoS One 2014;9:e107931. https://doi.org/10.1371/journal.pone.0107931 |
||||
| 151 Farrell AW, Halliday GM, Lyons JG: Brahma deficiency in keratinocytes promotes UV carcinogenesis by accelerating the escape from cell cycle arrest and the formation of DNA photolesions. J Dermatol Sci 2018;92:254-263. https://doi.org/10.1016/j.jdermsci.2018.11.006 |
||||
| 152 Bao X, Tang J, Lopez-Pajares V, Tao S, Qu K, Crabtree GR, Khavari PA: ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell 2013;12:193-203. https://doi.org/10.1016/j.stem.2012.12.014 |
||||
| 153 Saladi SV, Ross K, Karaayvaz M, Tata PR, Mou H, Rajagopal J, Ramaswamy S, Ellisen LW: ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017;31:35-49. https://doi.org/10.1016/j.ccell.2016.12.001 |
||||
| 154 Shrestha S, Adhikary G, Xu W, Kandasamy S, Eckert RL: ACTL6A suppresses p21(Cip1) expression to enhance the epidermal squamous cell carcinoma phenotype. Oncogene 2020;39:5855-5866. https://doi.org/10.1038/s41388-020-1371-8 |
||||
| 155 Piipponen M, Nissinen L, Kahari VM: Long non-coding RNAs in cutaneous biology and keratinocyte carcinomas. Cell Mol Life Sci 2020;77:4601-4614. https://doi.org/10.1007/s00018-020-03554-3 |
||||
| 156 Slack FJ, Chinnaiyan AM: The Role of Non-coding RNAs in Oncology. Cell 2019;179:1033-1055. https://doi.org/10.1016/j.cell.2019.10.017 |
||||
| 157 Chen P, Li C, Huang H, Liang L, Zhang J, Li Q, Wang Q, Zhang S, Zeng K, Zhang X, Liang J: Circular RNA profiles and the potential involvement of down-expression of hsa_circ_0001360 in cutaneous squamous cell carcinogenesis. FEBS Open Bio 2021;11:1209-1222. https://doi.org/10.1002/2211-5463.13114 |
||||
| 158 Das Mahapatra K, Pasquali L, Sondergaard JN, Lapins J, Nemeth IB, Baltas E, Kemeny L, Homey B, Moldovan LI, Kjems J, Kutter C, Sonkoly E, Kristensen LS, Pivarcsi A: A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci Rep 2020;10:3637. https://doi.org/10.1038/s41598-020-59660-6 |
||||
| 159 Inoue J, Fujiwara K, Hamamoto H, Kobayashi K, Inazawa J: Improving the Efficacy of EGFR Inhibitors by Topical Treatment of Cutaneous Squamous Cell Carcinoma with miR-634 Ointment. Mol Ther Oncolytics 2020;19:294-307. https://doi.org/10.1016/j.omto.2020.10.009 |
||||
| 160 Zhang M, Luo H, Hui L: MiR-3619-5p hampers proliferation and cisplatin resistance in cutaneous squamous-cell carcinoma via KPNA4. Biochem Biophys Res Commun 2019;513:419-425. https://doi.org/10.1016/j.bbrc.2019.03.203 |
||||
| 161 Rupaimoole R, Slack FJ: MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nature reviews Drug discovery 2017;16:203-222. https://doi.org/10.1038/nrd.2016.246 |
||||
| 162 Ross K: Towards topical microRNA-directed therapy for epidermal disorders. J Control Release 2018;269:136-147. https://doi.org/10.1016/j.jconrel.2017.11.013 |
||||
| 163 Depner S, Lederle W, Gutschalk C, Linde N, Zajonz A, Mueller MM: Cell type specific interleukin-6 induced responses in tumor keratinocytes and stromal fibroblasts are essential for invasive growth. Int J Cancer 2014;135:551-562. https://doi.org/10.1002/ijc.27951 |
||||
| 164 Commandeur S, Ho SH, de Gruijl FR, Willemze R, Tensen CP, El Ghalbzouri A: Functional characterization of cancer-associated fibroblasts of human cutaneous squamous cell carcinoma. Exp Dermatol 2011;20:737-742. https://doi.org/10.1111/j.1600-0625.2011.01305.x |
||||
| 165 Zhang J, Chen L, Xiao M, Wang C, Qin Z: FSP1+ fibroblasts promote skin carcinogenesis by maintaining MCP-1-mediated macrophage infiltration and chronic inflammation. Am J Pathol 2011;178:382-390. https://doi.org/10.1016/j.ajpath.2010.11.017 |
||||
| 166 Kim DE, Procopio MG, Ghosh S, Jo SH, Goruppi S, Magliozzi F, Bordignon P, Neel V, Angelino P, Dotto GP: Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation. J Exp Med 2017;214:2349-2368. https://doi.org/10.1084/jem.20170724 |
||||
| 167 Sobel K, Tham M, Stark HJ, Stammer H, Pratzel-Wunder S, Bickenbach JR, Boukamp P: Wnt-3a-activated human fibroblasts promote human keratinocyte proliferation and matrix destruction. Int J Cancer 2015;136:2786-2798. https://doi.org/10.1002/ijc.29336 |
||||
| 168 Katarkar A, Bottoni G, Clocchiatti A, Goruppi S, Bordignon P, Lazzaroni F, Gregnanin I, Ostano P, Neel V, Dotto GP: NOTCH1 gene amplification promotes expansion of Cancer Associated Fibroblast populations in human skin. Nat Commun 2020;11:5126. https://doi.org/10.1038/s41467-020-18919-2 |
||||
| 169 Bordignon P, Bottoni G, Xu X, Popescu AS, Truan Z, Guenova E, Kofler L, Jafari P, Ostano P, Rocken M, Neel V, Dotto GP: Dualism of FGF and TGF-beta Signaling in Heterogeneous Cancer-Associated Fibroblast Activation with ETV1 as a Critical Determinant. Cell Rep 2019;28:2358-2372 e2356. https://doi.org/10.1016/j.celrep.2019.07.092 |
||||
| 170 Tay SS, Roediger B, Tong PL, Tikoo S, Weninger W: The Skin-Resident Immune Network. Curr Dermatol Rep 2014;3:13-22. https://doi.org/10.1007/s13671-013-0063-9 |
||||
| 171 Khou S, Popa A, Luci C, Bihl F, Meghraoui-Kheddar A, Bourdely P, Salavagione E, Cosson E, Rubod A, Cazareth J, Barbry P, Mari B, Rezzonico R, Anjuere F, Braud VM: Tumor-Associated Neutrophils Dampen Adaptive Immunity and Promote Cutaneous Squamous Cell Carcinoma Development. Cancers (Basel) 2020;12:1860. https://doi.org/10.3390/cancers12071860 |
||||
| 172 Goncalves-Maia M, Gache Y, Basante M, Cosson E, Salavagione E, Muller M, Bernerd F, Avril MF, Schaub S, Sarasin A, Braud VM, Magnaldo T: NK Cell and Fibroblast-Mediated Regulation of Skin Squamous Cell Carcinoma Invasion by CLEC2A Is Compromised in Xeroderma Pigmentosum. J Invest Dermatol 2020;140:1723-1732. https://doi.org/10.1016/j.jid.2020.01.021 |
||||
| 173 Santos-Juanes J, Fernandez-Vega I, Lorenzo-Herrero S, Sordo-Bahamonde C, Martinez-Camblor P, Garcia-Pedrero JM, Vivanco B, Galache-Osuna C, Vazquez-Lopez F, Gonzalez S, Rodrigo JP: Lectin-like transcript 1 (LLT1) expression is associated with nodal metastasis in patients with head and neck cutaneous squamous cell carcinoma. Arch Dermatol Res 2019;311:369-376. https://doi.org/10.1007/s00403-019-01916-x |
||||
| 174 Jang TJ: Epithelial to mesenchymal transition in cutaneous squamous cell carcinoma is correlated with COX-2 expression but not with the presence of stromal macrophages or CD10-expressing cells. Virchows Arch 2012;460:481-487. https://doi.org/10.1007/s00428-012-1227-x |
||||
| 175 Pettersen JS, Fuentes-Duculan J, Suarez-Farinas M, Pierson KC, Pitts-Kiefer A, Fan L, Belkin DA, Wang CQ, Bhuvanendran S, Johnson-Huang LM, Bluth MJ, Krueger JG, Lowes MA, Carucci JA: Tumor-associated macrophages in the cutaneous SCC microenvironment are heterogeneously activated. J Invest Dermatol 2011;131:1322-1330. | ||||
| 176 Moussai D, Mitsui H, Pettersen JS, Pierson KC, Shah KR, Suarez-Farinas M, Cardinale IR, Bluth MJ, Krueger JG, Carucci JA: The human cutaneous squamous cell carcinoma microenvironment is characterized by increased lymphatic density and enhanced expression of macrophage-derived VEGF-C. J Invest Dermatol 2011;131:229-236. https://doi.org/10.1038/jid.2010.266 |
||||
| 177 Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM: Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol 2012;227:17-28. https://doi.org/10.1002/path.3989 |
||||
| 178 Minocha R, Martin AJ, Chen AC, Scolyer RA, Lyons JG, McKenzie CA, Madore J, Halliday GM, Damian DL: A Reduction in Inflammatory Macrophages May Contribute to Skin Cancer Chemoprevention by Nicotinamide. J Invest Dermatol 2019;139:467-469. https://doi.org/10.1016/j.jid.2018.08.018 |
||||
| 179 Bluth MJ, Zaba LC, Moussai D, Suarez-Farinas M, Kaporis H, Fan L, Pierson KC, White TR, Pitts-Kiefer A, Fuentes-Duculan J, Guttman-Yassky E, Krueger JG, Lowes MA, Carucci JA: Myeloid dendritic cells from human cutaneous squamous cell carcinoma are poor stimulators of T-cell proliferation. J Invest Dermatol 2009;129:2451-2462. https://doi.org/10.1038/jid.2009.96 |
||||
| 180 Fujita H, Suarez-Farinas M, Mitsui H, Gonzalez J, Bluth MJ, Zhang S, Felsen D, Krueger JG, Carucci JA: Langerhans cells from human cutaneous squamous cell carcinoma induce strong type 1 immunity. J Invest Dermatol 2012;132:1645-1655. https://doi.org/10.1038/jid.2012.34 |
||||
| 181 Munn DH: The host protecting the tumor from the host - targeting PDL1 expressed by host cells. J Clin Invest 2018;128:570-572. https://doi.org/10.1172/JCI99047 |
||||
| 182 Kamiya S, Kato J, Kamiya T, Yamashita T, Sumikawa Y, Hida T, Horimoto K, Sato S, Takahashi H, Sawada M, Kubo T, Torigoe T, Uhara H: Association between PD-L1 expression and lymph node metastasis in cutaneous squamous cell carcinoma. Asia Pac J Clin Oncol 2020;16:e108-e112. https://doi.org/10.1111/ajco.13102 |
||||
| 183 Garcia-Pedrero JM, Martinez-Camblor P, Diaz-Coto S, Munguia-Calzada P, Vallina-Alvarez A, Vazquez-Lopez F, Rodrigo JP, Santos-Juanes J: Tumor programmed cell death ligand 1 expression correlates with nodal metastasis in patients with cutaneous squamous cell carcinoma of the head and neck. J Am Acad Dermatol 2017;77:527-533. https://doi.org/10.1016/j.jaad.2017.05.047 |
||||
| 184 Varki V, Ioffe OB, Bentzen SM, Heath J, Cellini A, Feliciano J, Zandberg DP: PD-L1, B7-H3, and PD-1 expression in immunocompetent vs. immunosuppressed patients with cutaneous squamous cell carcinoma. Cancer Immunol Immunother 2018;67:805-814. https://doi.org/10.1007/s00262-018-2138-8 |
||||
| 185 Jiao Q, Liu C, Li W, Li W, Fang F, Qian Q, Zhang X: Programmed death-1 ligands 1 and 2 expression in cutaneous squamous cell carcinoma and their relationship with tumour- infiltrating dendritic cells. Clin Exp Immunol 2017;188:420-429. https://doi.org/10.1111/cei.12921 |
||||
| 186 Kraft S, Gadkaree SK, Deschler DG, Lin DT, Hoang MP, Emerick KS: Programmed cell death ligand-1 and cytotoxic T cell infiltrates in metastatic cutaneous squamous cell carcinoma of the head and neck. Head Neck 2020;42:3226-3234. https://doi.org/10.1002/hed.26370 |
||||
| 187 Slater NA, Googe PB: PD-L1 expression in cutaneous squamous cell carcinoma correlates with risk of metastasis. J Cutan Pathol 2016;43:663-670. https://doi.org/10.1111/cup.12728 |
||||
| 188 Lee A, Duggan S, Deeks ED: Cemiplimab: A Review in Advanced Cutaneous Squamous Cell Carcinoma. Drugs 2020;80:813-819. https://doi.org/10.1007/s40265-020-01302-2 |
||||
| 189 Grob JJ, Gonzalez R, Basset-Seguin N, Vornicova O, Schachter J, Joshi A, Meyer N, Grange F, Piulats JM, Bauman JR, Zhang P, Gumuscu B, Swaby RF, Hughes BGM: Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J Clin Oncol 2020:JCO1903054. https://doi.org/10.1200/JCO.19.03054 |
||||
| 190 Maubec E, Boubaya M, Petrow P, Beylot-Barry M, Basset-Seguin N, Deschamps L, Grob JJ, Dreno B, Scheer-Senyarich I, Bloch-Queyrat C, Leccia MT, Stefan A, Saiag P, Grange F, Meyer N, de Quatrebarbes J, Dinulescu M, Legoupil D, Machet L, Dereure O, et al.: Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients With Unresectable Cutaneous Squamous Cell Carcinomas. J Clin Oncol 2020;38:3051-3061. https://doi.org/10.1200/JCO.19.03357 |
||||
| 191 Ahmed SR, Petersen E, Patel R, Migden MR: Cemiplimab-rwlc as first and only treatment for advanced cutaneous squamous cell carcinoma. Expert Rev Clin Pharmacol 2019;12:947-951. https://doi.org/10.1080/17512433.2019.1665026 |
||||
| 192 Mittapalli VR, Madl J, Loffek S, Kiritsi D, Kern JS, Romer W, Nystrom A, Bruckner-Tuderman L: Injury-Driven Stiffening of the Dermis Expedites Skin Carcinoma Progression. Cancer Res 2016;76:940-951. https://doi.org/10.1158/0008-5472.CAN-15-1348 |
||||
| 193 van Kempen LC, Rijntjes J, Claes A, Blokx WA, Gerritsen MJ, Ruiter DJ, van Muijen GN: Type I collagen synthesis parallels the conversion of keratinocytic intraepidermal neoplasia to cutaneous squamous cell carcinoma. J Pathol 2004;204:333-339. https://doi.org/10.1002/path.1659 |
||||
| 194 Fiore VF, Krajnc M, Quiroz FG, Levorse J, Pasolli HA, Shvartsman SY, Fuchs E: Mechanics of a multilayer epithelium instruct tumour architecture and function. Nature 2020;585:433-439. https://doi.org/10.1038/s41586-020-2695-9 |
||||
| 195 Desgrosellier JS, Cheresh DA: Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010;10:9-22. https://doi.org/10.1038/nrc2748 |
||||
| 196 Reuter JA, Ortiz-Urda S, Kretz M, Garcia J, Scholl FA, Pasmooij AM, Cassarino D, Chang HY, Khavari PA: Modeling inducible human tissue neoplasia identifies an extracellular matrix interaction network involved in cancer progression. Cancer Cell 2009;15:477-488. https://doi.org/10.1016/j.ccr.2009.04.002 |
||||
| 197 Kariya Y, Oyama M, Hashimoto Y, Gu J, Kariya Y: beta4-Integrin/PI3K Signaling Promotes Tumor Progression through the Galectin-3-N-Glycan Complex. Mol Cancer Res 2018;16:1024-1034. https://doi.org/10.1158/1541-7786.MCR-17-0365 |
||||
| 198 Lincoln V, Chao L, Woodley DT, Murrell D, Kim M, O'Toole EA, Ly A, Cogan J, Mosallaei D, Wysong A, Chen M: Over-Expression of stromal periostin correlates with poor Prognosis of cutaneous squamous Cell carcinomas. Exp Dermatol 2021:30:698-704. https://doi.org/10.1111/exd.14281 |
||||
| 199 Caley MP, Martins VL, Moore K, Lashari M, Nissinen L, Kahari VM, Alexander S, Jones E, Harwood CA, Jones J, Donaldson M, Marshall JF, O'Toole EA: Loss of Laminin alpha3 induces cell invasion and macrophage infiltration in cutaneous squamous cell carcinoma. Br J Dermatol 2020;184:923-934. https://doi.org/10.1111/bjd.19471 |
||||
| 200 Siljamaki E, Rappu P, Riihila P, Nissinen L, Kahari VM, Heino J: H-Ras activation and fibroblast-induced TGF-beta signaling promote laminin-332 accumulation and invasion in cutaneous squamous cell carcinoma. Matrix Biol 2020;87:26-47. https://doi.org/10.1016/j.matbio.2019.09.001 |
||||
| 201 Roh MR, Zheng Z, Kim HS, Kwon JE, Jeung HC, Rha SY, Chung KY: Differential expression patterns of MMPs and their role in the invasion of epithelial premalignant tumors and invasive cutaneous squamous cell carcinoma. Exp Mol Pathol 2012;92:236-242. https://doi.org/10.1016/j.yexmp.2012.01.003 |
||||
| 202 O'Grady A, Dunne C, O'Kelly P, Murphy GM, Leader M, Kay E: Differential expression of matrix metalloproteinase (MMP)-2, MMP-9 and tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 in non-melanoma skin cancer: implications for tumour progression. Histopathology 2007;51:793-804. https://doi.org/10.1111/j.1365-2559.2007.02885.x |
||||
| 203 Lee JH, Piao MS, Choi JY, Yun SJ, Lee JB, Lee SC: Up-regulation of cyclooxygenase 2 and matrix metalloproteinases-2 and -9 in cutaneous squamous cell carcinoma: active role of inflammation and tissue remodeling in carcinogenesis. Ann Dermatol 2013;25:145-151. https://doi.org/10.5021/ad.2013.25.2.145 |
||||
| 204 Mitsui H, Suarez-Farinas M, Gulati N, Shah KR, Cannizzaro MV, Coats I, Felsen D, Krueger JG, Carucci JA: Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol 2014;134:1418-1427. https://doi.org/10.1038/jid.2013.494 |
||||
| 205 Ahmed Haji Omar A, Haglund C, Virolainen S, Hayry V, Atula T, Kontio R, Salo T, Sorsa T, Hagstrom J: MMP-7, MMP-8, and MMP-9 in oral and cutaneous squamous cell carcinomas. Oral Surg Oral Med Oral Pathol Oral Radiol 2015;119:459-467. https://doi.org/10.1016/j.oooo.2014.12.019 |
||||
| 206 Kadeh H, Saravani S, Heydari F, Shahraki S: Differential immunohistochemical expression of matrix metalloproteinase-10 (MMP-10) in non-melanoma skin cancers of the head and neck. Pathol Res Pract 2016;212:867-871. https://doi.org/10.1016/j.prp.2016.06.015 |
||||
| 207 Fundyler O, Khanna M, Smoller BR: Metalloproteinase-2 expression correlates with aggressiveness of cutaneous squamous cell carcinomas. Mod Pathol 2004;17:496-502. https://doi.org/10.1038/modpathol.3800066 |
||||
| 208 Sweeny L, Dean NR, Frederick JW, Magnuson JS, Carroll WR, Desmond RA, Rosenthal EL: CD147 expression in advanced cutaneous squamous cell carcinoma. J Cutan Pathol 2012;39:603-609. https://doi.org/10.1111/j.1600-0560.2012.01912.x |
||||
| 209 Fields GB: Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front Immunol 2019;10:1278. https://doi.org/10.3389/fimmu.2019.01278 |
||||
| 210 Gao L, Jin HJ, Zhang D, Lin Q: Silencing circRNA_001937 may inhibit cutaneous squamous cell carcinoma proliferation and induce apoptosis by preventing the sponging of the miRNA5973p/FOSL2 pathway. Int J Mol Med 2020;46:1653-1660. https://doi.org/10.3892/ijmm.2020.4723 |
||||
| 211 An X, Liu X, Ma G, Li C: Upregulated circular RNA circ_0070934 facilitates cutaneous squamous cell carcinoma cell growth and invasion by sponging miR-1238 and miR-1247-5p. Biochem Biophys Res Commun 2019;513:380-385. https://doi.org/10.1016/j.bbrc.2019.04.017 |
||||
| 212 Zhang DW, Wu HY, Zhu CR, Wu DD: CircRNA hsa_circ_0070934 functions as a competitive endogenous RNA to regulate HOXB7 expression by sponging miR12363p in cutaneous squamous cell carcinoma. Int J Oncol 2020;57:478-487. https://doi.org/10.3892/ijo.2020.5066 |
||||
| 213 Lu X, Gan Q, Gan C: Circular RNA circSEC24A Promotes Cutaneous Squamous Cell Carcinoma Progression by Regulating miR-1193/MAP3K9 Axis. Onco Targets Ther 2021;14:653-666. https://doi.org/10.2147/OTT.S275691 |
||||
| 214 Mei XL, Zhong S: Long noncoding RNA LINC00520 prevents the progression of cutaneous squamous cell carcinoma through the inactivation of the PI3K/Akt signaling pathway by downregulating EGFR. Chin Med J (Engl) 2019;132:454-465. https://doi.org/10.1097/CM9.0000000000000070 |
||||
| 215 Lee CS, Mah A, Aros CJ, Lopez-Pajares V, Bhaduri A, Webster DE, Kretz M, Khavari PA: Cancer-Associated Long Noncoding RNA SMRT-2 Controls Epidermal Differentiation. J Invest Dermatol 2018;138:1445-1449. https://doi.org/10.1016/j.jid.2018.01.003 |
||||
| 216 Zhou W, Zhang S, Li J, Li Z, Wang Y, Li X: lncRNA TINCR participates in ALA-PDT-induced apoptosis and autophagy in cutaneous squamous cell carcinoma. J Cell Biochem 2019;120:13893-13902. https://doi.org/10.1002/jcb.28662 |
||||
| 217 Lu D, Sun L, Li Z, Mu Z: lncRNA EZRAS1 knockdown represses proliferation, migration and invasion of cSCC via the PI3K/AKT signaling pathway. Mol Med Rep 2021;23:76. https://doi.org/10.3892/mmr.2020.11714 |
||||
| 218 Yu GJ, Sun Y, Zhang DW, Zhang P: Long non-coding RNA HOTAIR functions as a competitive endogenous RNA to regulate PRAF2 expression by sponging miR-326 in cutaneous squamous cell carcinoma. Cancer Cell Int 2019;19:270. https://doi.org/10.1186/s12935-019-0992-x |
||||
| 219 Li F, Liao J, Duan X, He Y, Liao Y: Upregulation of LINC00319 indicates a poor prognosis and promotes cell proliferation and invasion in cutaneous squamous cell carcinoma. J Cell Biochem 2018;119:10393-10405. https://doi.org/10.1002/jcb.27388 |
||||
| 220 Wang J, Li C, Xu L, Yang C, Zhang X: MiR-1193 was sponged by LINC00963 and inhibited cutaneous squamous cell carcinoma progression by targeting SOX4. Pathol Res Pract 2019;215:152600. https://doi.org/10.1016/j.prp.2019.152600 |
||||
| 221 Zhang Y, Gao L, Ma S, Ma J, Wang Y, Li S, Hu X, Han S, Zhou M, Zhou L, Ding Z: MALAT1-KTN1-EGFR regulatory axis promotes the development of cutaneous squamous cell carcinoma. Cell Death Differ 2019;26:2061-2073. https://doi.org/10.1038/s41418-019-0288-7 |
||||
| 222 Piipponen M, Nissinen L, Farshchian M, Riihila P, Kivisaari A, Kallajoki M, Peltonen J, Peltonen S, Kahari VM: Long Noncoding RNA PICSAR Promotes Growth of Cutaneous Squamous Cell Carcinoma by Regulating ERK1/2 Activity. J Invest Dermatol 2016;136:1701-1710. https://doi.org/10.1016/j.jid.2016.03.028 |
||||
| 223 Lu X, Gan Q, Gan C, Zheng Y, Cai B, Li X, Li D, Yin G: Long non-coding RNA PICSAR knockdown inhibits the progression of cutaneous squamous cell carcinoma by regulating miR-125b/YAP1 axis. Life Sci 2020:118303. https://doi.org/10.1016/j.lfs.2020.118303 |
||||
| 224 Piipponen M, Nissinen L, Riihila P, Farshchian M, Kallajoki M, Peltonen J, Peltonen S, Kahari VM: p53-Regulated Long Noncoding RNA PRECSIT Promotes Progression of Cutaneous Squamous Cell Carcinoma via STAT3 Signaling. Am J Pathol 2020;190:503-517. https://doi.org/10.1016/j.ajpath.2019.10.019 |
||||
| 225 Zhang Z, Jia M, Wen C, He A, Ma Z: LncRNA SCARNA2 induces cutaneous squamous cell carcinoma progression via modulating miR-342-3p expression. J Gene Med 2020:e3242. https://doi.org/10.1002/jgm.3242 |
||||
| 226 Zhou J, Liu R, Luo C, Zhou X, Xia K, Chen X, Zhou M, Zou Q, Cao P, Cao K: MiR-20a inhibits cutaneous squamous cell carcinoma metastasis and proliferation by directly targeting LIMK1. Cancer Biol Ther 2014;15:1340-1349. https://doi.org/10.4161/cbt.29821 |
||||
| 227 Zhang L, Xiang P, Han X, Wu L, Li X, Xiong Z: Decreased expression of microRNA-20a promotes tumor progression and predicts poor prognosis of cutaneous squamous cell carcinoma. Int J Clin Exp Pathol 2015;8:11446-11451. | ||||
| 228 Zheng W, Li ZY, Zhao DL, Li XL, Liu R: microRNA-26a Directly Targeting MMP14 and MMP16 Inhibits the Cancer Cell Proliferation, Migration and Invasion in Cutaneous Squamous Cell Carcinoma. Cancer Manag Res 2020;12:7087-7095. https://doi.org/10.2147/CMAR.S265775 |
||||
| 229 Wang Y, Deng X, Dai Y, Niu X, Zhou M: miR-27a Downregulation Promotes Cutaneous Squamous Cell Carcinoma Progression via Targeting EGFR. Front Oncol 2019;9:1565. https://doi.org/10.3389/fonc.2019.01565 |
||||
| 230 Shao J, Liang J, Zhong S: miR-30a-5p modulates traits of cutaneous squamous cell carcinoma (cSCC) via forkhead box protein G1 (FOXG1). Neoplasma 2019;66:908-917. https://doi.org/10.4149/neo_2018_181205N923 |
||||
| 231 Lefort K, Brooks Y, Ostano P, Cario-Andre M, Calpini V, Guinea-Viniegra J, Albinger-Hegyi A, Hoetzenecker W, Kolfschoten I, Wagner EF, Werner S, Dotto GP: A miR-34a-SIRT6 axis in the squamous cell differentiation network. EMBO J 2013;32:2248-2263. https://doi.org/10.1038/emboj.2013.156 |
||||
| 232 Li S, Luo C, Zhou J, Zhang Y: MicroRNA-34a directly targets high-mobility group box 1 and inhibits the cancer cell proliferation, migration and invasion in cutaneous squamous cell carcinoma. Exp Ther Med 2017;14:5611-5618. https://doi.org/10.3892/etm.2017.5245 |
||||
| 233 Antonini D, Russo MT, De Rosa L, Gorrese M, Del Vecchio L, Missero C: Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. J Invest Dermatol 2010;130:1249-1257. https://doi.org/10.1038/jid.2009.438 |
||||
| 234 Yamane K, Jinnin M, Etoh T, Kobayashi Y, Shimozono N, Fukushima S, Masuguchi S, Maruo K, Inoue Y, Ishihara T, Aoi J, Oike Y, Ihn H: Down-regulation of miR-124/-214 in cutaneous squamous cell carcinoma mediates abnormal cell proliferation via the induction of ERK. J Mol Med (Berl) 2013;91:69-81. https://doi.org/10.1007/s00109-012-0935-7 |
||||
| 235 Xu N, Zhang L, Meisgen F, Harada M, Heilborn J, Homey B, Grander D, Stahle M, Sonkoly E, Pivarcsi A: MicroRNA-125b down-regulates matrix metallopeptidase 13 and inhibits cutaneous squamous cell carcinoma cell proliferation, migration, and invasion. J Biol Chem 2012;287:29899-29908. https://doi.org/10.1074/jbc.M112.391243 |
||||
| 236 Tian K, Liu W, Zhang J, Fan X, Liu J, Zhao N, Yao C, Miao G: MicroRNA-125b exerts antitumor functions in cutaneous squamous cell carcinoma by targeting the STAT3 pathway. Cell Mol Biol Lett 2020;25:12. https://doi.org/10.1186/s11658-020-00207-y |
||||
| 237 Luo Q, Li W, Zhao T, Tian X, Liu Y, Zhang X: Role of miR-148a in cutaneous squamous cell carcinoma by repression of MAPK pathway. Arch Biochem Biophys 2015;583:47-54. https://doi.org/10.1016/j.abb.2015.07.022 |
||||
| 238 Neu J, Dziunycz PJ, Dzung A, Lefort K, Falke M, Denzler R, Freiberger SN, Iotzova-Weiss G, Kuzmanov A, Levesque MP, Dotto GP, Hofbauer GFL: miR-181a decelerates proliferation in cutaneous squamous cell carcinoma by targeting the proto-oncogene KRAS. PLoS One 2017;12:e0185028. https://doi.org/10.1371/journal.pone.0185028 |
||||
| 239 Wang SH, Zhou JD, He QY, Yin ZQ, Cao K, Luo CQ: MiR-199a inhibits the ability of proliferation and migration by regulating CD44-Ezrin signaling in cutaneous squamous cell carcinoma cells. Int J Clin Exp Pathol 2014;7:7131-7141. | ||||
| 240 Lu RH, Xiao ZQ, Zhou JD, Yin CQ, Chen ZZ, Tang FJ, Wang SH: MiR-199a-5p represses the stemness of cutaneous squamous cell carcinoma stem cells by targeting Sirt1 and CD44ICD cleavage signaling. Cell Cycle (Georgetown, Tex) 2020;19:1-14. https://doi.org/10.1080/15384101.2019.1689482 |
||||
| 241 Kim BK, Kim I, Yoon SK: Identification of miR-199a-5p target genes in the skin keratinocyte and their expression in cutaneous squamous cell carcinoma. J Dermatol Sci 2015;79:137-147. https://doi.org/10.1016/j.jdermsci.2015.05.005 |
||||
| 242 Canueto J, Cardenoso-Alvarez E, Garcia-Hernandez JL, Galindo-Villardon P, Vicente-Galindo P, Vicente-Villardon JL, Alonso-Lopez D, De Las Rivas J, Valero J, Moyano-Sanz E, Fernandez-Lopez E, Mao JH, Castellanos-Martin A, Roman-Curto C, Perez-Losada J: MicroRNA (miR)-203 and miR-205 expression patterns identify subgroups of prognosis in cutaneous squamous cell carcinoma. Br J Dermatol 2017;177:168-178. https://doi.org/10.1111/bjd.15236 |
||||
| 243 Toll A, Salgado R, Espinet B, Diaz-Lagares A, Hernandez-Ruiz E, Andrades E, Sandoval J, Esteller M, Pujol RM, Hernandez-Munoz I: MiR-204 silencing in intraepithelial to invasive cutaneous squamous cell carcinoma progression. Mol Cancer 2016;15:53. https://doi.org/10.1186/s12943-016-0537-z |
||||
| 244 Robinson DJ, Patel A, Purdie KJ, Wang J, Rizvi H, Hufbauer M, Ostano P, Akgul B, Chiorino G, Harwood CA, Bergamaschi D: Epigenetic Regulation of iASPP-p63 Feedback Loop in Cutaneous Squamous Cell Carcinoma. J Invest Dermatol 2019;139:1658-1671 e1658. https://doi.org/10.1016/j.jid.2019.01.020 |
||||
| 245 Ma X, Wu D, Zhang X, Shao X, Hu G: microRNA-214 Prevents Traits of Cutaneous Squamous Cell Carcinoma via VEGFA and Bcl-2. Technol Cancer Res Treat 2020;19:1533033820980098. https://doi.org/10.1177/1533033820980098 |
||||
| 246 Feng C, Zhang HL, Zeng A, Bai M, Wang XJ: Tumor-Suppressive MicroRNA-216b Binds to TPX2, Activating the p53 Signaling in Human Cutaneous Squamous Cell Carcinoma. Mol Ther Nucleic Acids 2020;20:186-195. https://doi.org/10.1016/j.omtn.2020.01.022 |
||||
| 247 Fu J, Zhao J, Zhang H, Fan X, Geng W, Qiao S: MicroRNA-451a prevents cutaneous squamous cell carcinoma progression via the 3-phosphoinositide-dependent protein kinase-1-mediated PI3K/AKT signaling pathway. Exp Ther Med 2021;21:116. https://doi.org/10.3892/etm.2020.9548 |
||||
| 248 Mizrahi A, Barzilai A, Gur-Wahnon D, Ben-Dov IZ, Glassberg S, Meningher T, Elharar E, Masalha M, Jacob-Hirsch J, Tabibian-Keissar H, Barshack I, Roszik J, Leibowitz-Amit R, Sidi Y, Avni D: Alterations of microRNAs throughout the malignant evolution of cutaneous squamous cell carcinoma: the role of miR-497 in epithelial to mesenchymal transition of keratinocytes. Oncogene 2018;37:218-230. https://doi.org/10.1038/onc.2017.315 |
||||
| 249 Wei XH, Gu XL, Zhou XT, Ma M, Lou CX: miR-497 promotes the progression of cutaneous squamous cell carcinoma through FAM114A2. Eur Rev Med Pharmacol Sci 2018;22:7348-7355. | ||||
| 250 Xiong Z, Jiang B, Li G: Downregulation of miR-10a inhibits cutaneous squamous cell carcinoma cell proliferation, migration, and invasion by targeting Syndecan-1. Int J Clin Exp Pathol 2020;13:2502-2512. | ||||
| 251 Yin S, Lin X: MicroRNA-21 Contributes to Cutaneous Squamous Cell Carcinoma Progression via Mediating TIMP3/PI3K/AKT Signaling Axis. Int J Gen Med 2021;14:27-39. https://doi.org/10.2147/IJGM.S275016 |
||||
| 252 Wang A, Landen NX, Meisgen F, Lohcharoenkal W, Stahle M, Sonkoly E, Pivarcsi A: MicroRNA-31 is overexpressed in cutaneous squamous cell carcinoma and regulates cell motility and colony formation ability of tumor cells. PLoS One 2014;9:e103206. https://doi.org/10.1371/journal.pone.0103206 |
||||
| 253 Lin N, Zhou Y, Lian X, Tu Y: MicroRNA-31 functions as an oncogenic microRNA in cutaneous squamous cell carcinoma cells by targeting RhoTBT1. Oncol Lett 2017;13:1078-1082. https://doi.org/10.3892/ol.2017.5554 |
||||
| 254 Bai X, Zhou Y, Chen P, Yang M, Xu J: MicroRNA-142-5p induces cancer stem cell-like properties of cutaneous squamous cell carcinoma via inhibiting PTEN. J Cell Biochem 2018;119:2179-2188. https://doi.org/10.1002/jcb.26379 |
||||
| 255 Tian J, Shen R, Yan Y, Deng L: miR-186 promotes tumor growth in cutaneous squamous cell carcinoma by inhibiting apoptotic protease activating factor-1. Exp Ther Med 2018;16:4010-4018. https://doi.org/10.3892/etm.2018.6679 |
||||
| 256 Hu X, Liu Y, Ai P, He S, Liu L, Chen C, Tan Y, Wang T: MicroRNA-186 promotes cell proliferation and inhibits cell apoptosis in cutaneous squamous cell carcinoma by targeting RETREG1. Exp Ther Med 2019;17:1930-1938. https://doi.org/10.3892/etm.2019.7154 |
||||
| 257 Ory B, Ramsey MR, Wilson C, Vadysirisack DD, Forster N, Rocco JW, Rothenberg SM, Ellisen LW: A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. J Clin Invest 2011;121:809-820. https://doi.org/10.1172/JCI43897 |
||||
| 258 Bai M, Zhang M, Long F, Yu N, Zeng A, Wang X: MiR-217 promotes cutaneous squamous cell carcinoma progression by targeting PTRF. Am J Transl Res 2017;9:647-655. | ||||
| 259 Gong ZH, Zhou F, Shi C, Xiang T, Zhou CK, Wang QQ, Jiang YS, Gao SF: miRNA-221 promotes cutaneous squamous cell carcinoma progression by targeting PTEN. Cell Mol Biol Lett 2019;24:9. https://doi.org/10.1186/s11658-018-0131-z |
||||
| 260 Chen B, Pan W, Lin X, Hu Z, Jin Y, Chen H, Ma G, Qiu Y, Chang L, Hua C, Zou Y, Gao Y, Ying H, Lv D: MicroRNA-346 functions as an oncogene in cutaneous squamous cell carcinoma. Tumour Biol 2016;37:2765-2771. https://doi.org/10.1007/s13277-015-4046-2 |
||||
| 261 Zhou J, Zhang Y, Han Z, Dong Z, Cao T, Wei A, Guo P, Meng Q: miR-506 contributes to malignancy of cutaneous squamous cell carcinoma via targeting of P65 and LAMC1. Cell cycle (Georgetown, Tex) 2019;18:333-345. https://doi.org/10.1080/15384101.2019.1568747 |
||||
| 262 Liu P, Shi L, Ding Y, Luan J, Shan X, Li Q, Zhang S: MicroRNA-766 Promotes The Proliferation, Migration And Invasion, And Inhibits The Apoptosis Of Cutaneous Squamous Cell Carcinoma Cells By Targeting PDCD5. Onco Targets Ther 2020;13:4099-4110. https://doi.org/10.2147/OTT.S222821 |
||||
| 263 Zhou M, Liu W, Ma S, Cao H, Peng X, Guo L, Zhou X, Zheng L, Guo L, Wan M, Shi W, He Y, Lu C, Jiang L, Ou C, Guo Y, Ding Z: A novel onco-miR-365 induces cutaneous squamous cell carcinoma. Carcinogenesis 2013;34:1653-1659. https://doi.org/10.1093/carcin/bgt097 |
||||
| 264 Zhou L, Gao R, Wang Y, Zhou M, Ding Z: Loss of BAX by miR-365 Promotes Cutaneous Squamous Cell Carcinoma Progression by Suppressing Apoptosis. Int J Mol Sci 2017;18:1157. https://doi.org/10.3390/ijms18061157 |
||||