×
![]()
Corresponding Author: Karl Kunzelmann
Institut für Physiologie, Universität Regensburg,
Universitätsstraße 31, 93053 Regensburg (Germany)
Tel. +49 (0)941 943 4302, Fax +49 (0)941 943 4315 , E-Mail karl.kunzelmann@ur.de
P. aeruginosa Induced Lipid Peroxidation Causes Ferroptotic Cell Death in Airways
Jiraporn Ousingsawata Rainer Schreibera Erich Gulbinsb Markus Kamlerc Karl Kunzelmanna
aInstitut für Physiologie, Universität Regensburg, Regensburg, Germany, bInstitute of Molecular Biology, University of Duisburg-Essen, Medical School Essen, Essen, Germany, cDepartment of Thoracic and Cardiovascular Surgery, Division of Thoracic Transplantation, University Hospital Essen, Essen, Germany
Introduction
Oxidative stress and infections by P. aeruginosa play prominent roles in the lung pathology of cystic fibrosis (CF). Lungs of mice lacking expression of CFTR exhibit a redox imbalance in the airway surface liquid (ASL), leading to higher levels of endogenous reactive oxygen species (ROS) and lipid peroxidation [1]. Moreover, pulmonary accumulation of ceramide has been observed in CFTR-deficient mice, causing constitutive age-dependent pulmonary inflammation and high susceptibility to severe P. aeruginosa infections [2]. Further evidence for lipid peroxides in CF human airways has been found along with inflammasome-dependent inflammation, which contributes to respiratory infections [3, 4]. Another study, however, did not find inflammasome-activation by P. aeruginosa isolates [5].
Dar et al. reported expression of lipoxygenase (pLoxA) by P. aeruginosa. By means of pLoxA, host arachidonic acid–phosphatidylethanolamines (AA-PE) are oxidized and ferroptotic cell death is triggered in human bronchial epithelial cells. Elevated levels of oxidized AA-PE were detected in airway samples from P. aeruginosa-infected CF patients, but not in uninfected CF lungs or lungs infected with other pathogens such as Burkholderia cenocepacia, Achromobacter xylosoxidans, or Mycobacterium abscessus [6]. We previously examined ROS-mediated cell death in airway and intestinal epithelium. In contrast to an earlier study that suggested release of the ROS-scavenger glutathione (GSH) by CFTR, thus enhancing cytosolic ROS [7], we found identical ROS-levels in CFTR+/+ vs. CFTR-/- cells [8]. The phospholipid scramblase and ion channel TMEM16F was found to contribute to ferroptotic cell death [8]. Spontaneous cell death was massively reduced in the intestine of TMEM16F knockout mice, and was also observed after knockout of TMEM16F in ciliated airway epithelial cells [9]. These and other data [10] strongly suggested a role of TMEM16F in ferroptosis.
Along the same line we found that TMEM16F and the Ca2+ activated Cl- channel TMEM16A act cooperatively on the activation of ion currents and induction of cell death. Moreover, both TMEM16A and TMEM16F are activated through lipid peroxidation and other plasma membrane lipid modifications [11]. Because activation of TMEM16A by lipid peroxidation was shown to drive growth of renal cysts in vitro, we proposed inhibition of TMEM16A or inhibition of lipid peroxidation / ferroptosis as a therapeutic strategy, to delay cyst development in polycystic kidney disease [12]. Here we examined how lipid peroxidation in human and mouse airways correlates with bacterial infection and the presence of a CF-phenotype. In vitro analysis of airway epithelial cells suggests a role TMEM16A and TMEM16F in ferroptosis and suggests inhibitors of lipid peroxidation / ferroptosis to reduce airway epithelial cell death in cystic fibrosis [13].
Materials and Methods
Tissue preparation from human, piglet, and mouse lung
Lung tissues were obtained from 5 non-cystic fibrosis (non-CF) and from 5 cystic fibrosis patients (CF). The analysis of human lung sections has been approved by the Ethikkommission des Universitätsklinikum Essen (West German Heart and Vascular Center Essen, University of Duisburg-Essen, Hufelandstrasse 55, 45122 Essen, Germany), Genehmigungsnummer 17-7326-BO. The wild type CFTR (CFTR+/+) and knockout CFTR (CFTR-/-) piglet were generated in Prof. Dr. Klymiuk’s laboratory (Institute of Molecular Animal Breeding and Biotechnology, Gene Center, Ludwig-Maximilians-Universität München), who kindly provided the piglet lung tissue samples [14]. Piglet lungs were fixed in 4% paraformaldehyde in PBS, dehydrated with ethanol to xylene gradient and embedded in paraffin. Cftr+/+ and Cftr-/- mice were infected with Pseudomonas aeruginosa (P.ae) or Staphylococcus aureus (S.au). Growth of P. aeruginosa and S. aureus and treatment of animals has been described in detail previously [15].
Cells and treatment
Human cystic fibrosis bronchial epithelial cell lines stably expressing wtCFTR (CFBE/wtCFTR) or F508del‐CFTR (CFBE/F508del-CFTR) were cultured in MEM media supplemented with 10% heat-inactivated fetal calf serum, 2mM L-glutamine and 2.5 μg/ml puromycin (All culture media and supplements were from Capricorn Scientific, Ebsdorfergrund, Germany). Cells were grown at 37°C in a humidified atmosphere with 5% (v/v) CO2. For P. aeruginosa treatment, the frozen bacteria stock was plated from fresh tryptic soy agar plate (TSA; Becton Dickinson) and grown at 37°C for 14 to 16 hr. P. aeruginosa were resuspended in trypticase soy broth (Becton Dickinson) and then incubated at 37°C for 1 hr with shaking at 130 rpm to obtain bacteria in the early logarithmic growth phase. Bacteria were then washed and resuspended in warmed cell culture medium containing with 10 mM HEPES. The final concentration of bacteria was quantified by spectrophotometry. All inhibitors and compounds were applied to cells 2 h before P. aeruginosa treatment.
siRNA-knockdown of ANO1 (TMEM16A) and ANO6 (TMEM16F)
Knockdown of TMEM16 proteins in CFBE cells was performed through transfection by electroporation of siTMEM16A or siTMEM16F (each 1 µg/ml; both from Invitrogen, Carlsbad, CA, USA), using a neon transfection system (Invitrogen, Carlsbad, CA, USA) with a program of 3 pulses, 1650 V, 10 ms. Alternatively, the siRNAs were transfected using standard protocols for Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). Scrambled siRNA (Invitrogen, Carlsbad, CA, USA) was transfected as control. All cells transfected with siRNA were used for experiments 72 h after transfection. Successful knockdown was verified by Western-blotting, which indicated a complete knockdown for both ANO1 and ANO6.
Immunocytochemistry of 4HNE in tissue and cells
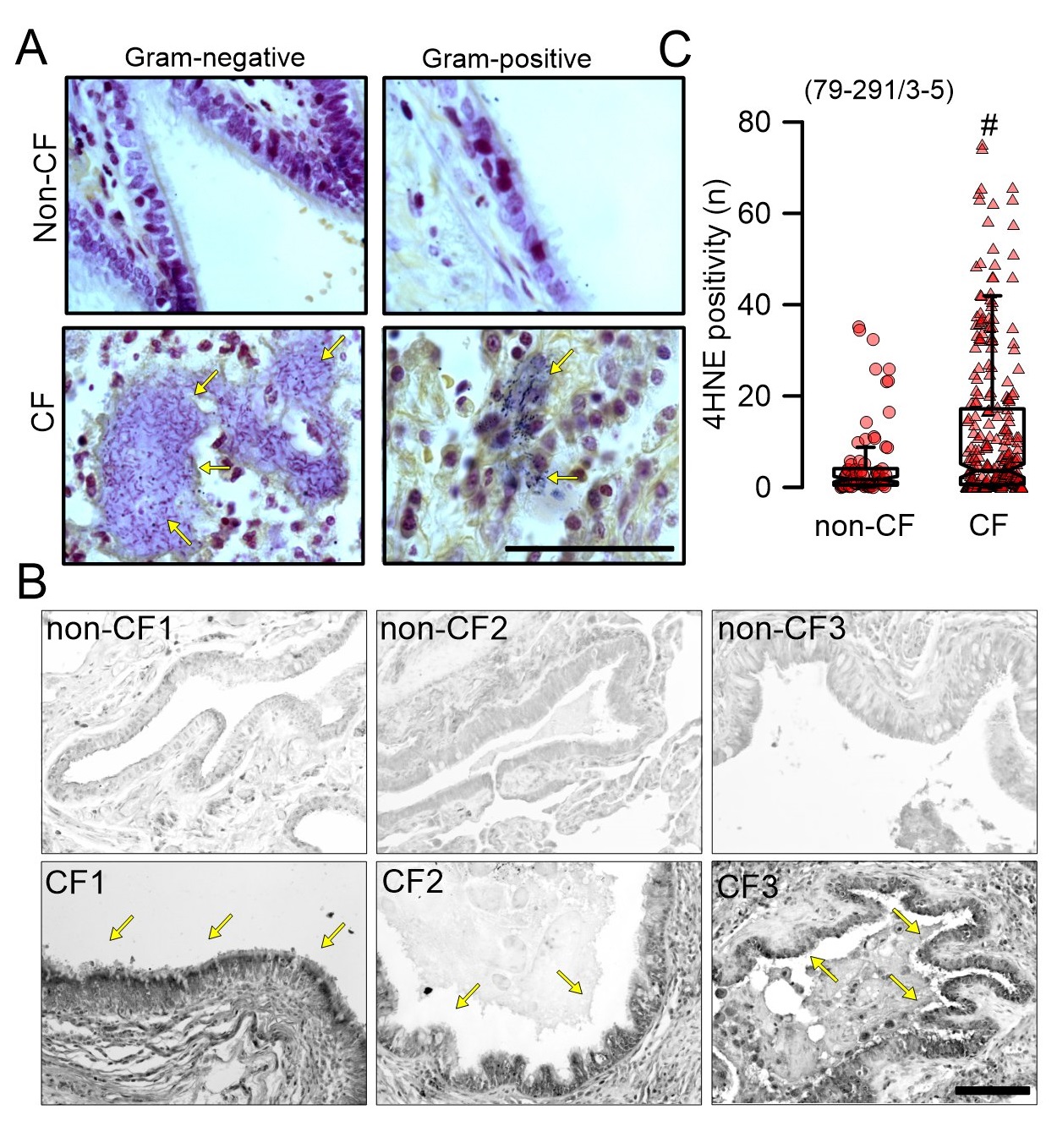
Lipid peroxidation in lung tissue sections was detect by 4-Hydroxy-2-nonenal (4HNE) staining. Briefly, paraffin embedded human lung sections (5 µm) were deparaffinised, rehydrated and then incubated in sodium citrate buffer pH 6.0 at 95°C for 15 min. Quenching of endogenous peroxidase was performed with 3%H2O2 for 15 min before incubation with rabbit anti-4HNE antibody (Alpha Diagnostic International, Tx, USA) overnight at 4°C. After washing, biotinylated donkey anti-rabbit IgG (Santa Cruz, Heidelberg, Germany) was applied for 1 h at room temperature. Sections were washed and then incubated with avidin-peroxidase complex (Vectastain kit, Vector laboratories) for 1.5 h at room temperature. The peroxidase was then developed by DAB (Sigma-Aldrich, Taufkirchen, Germany). Stainings were assessed by light microscopy, and were analysed using ImageJ. To analyse lipid peroxidation in CFBE human airway epithelial cells, CFBE cells were grown on glass cover slips and incubated with P. aeruginosa for 0, 2 and 4 h. Cells were fixed with 4% paraformaldehyde 15 min. After washing in PBS, cells were incubated with primary antibody rabbit anti 4HNE (Alpha Diagnostic International, Tx, USA) at 4°C overnight. Cells were incubated with secondary AlexaFluro 488 goat anti-rabbit IgG (Molecular Probes, Thermo Fisher, Germany) for 1h at room temperature. After washing with PBS, cells were counterstained with Hoechst33342 (Sigma-Aldrich, Taufkirchen, Germany) and mounted with DakoCytomation fluorescent mounting media (DAKO cytomation). Immunofluorescence was detected using an Axio Observer microscope equipped with Axiocams 503 mono and ApoTome.2. Immunofluorescence signals from 4HNE stainings in the airway epithelium were determined by marking the regions of interest (ROI; epithelium only) and by setting the background brightness as reference. The reference was kept throughout the entire analysis of all images. A large number of images (n=79-291) was analyzed from lungs of each patient. Microscopic setting for excitation, exposure etc. were kept constant throughout the entire analysis. Values were detected by using Axio Observer microscope.
Detection of intracellular ROS and cell death by propidium iodide
Intracellular ROS was detected using Fluorometric intracellular ROS kit (Deep red fluorescence, Sigma, Taufkirchen, Germany). The unbiased tracking software of ZEN 3.0 automatically detected and counted fluorescent cells. Microscopic settings were identical throughout the entire analysis of all images. Data are shown as number of cells divided by total number of cells counted. Cell death was detected by propidium iodide permeabilization. Fluorescence was detected using Axio Observer microscope and ZEN 3.0 (blue edition) software (Zeiss, Oberkochen, Germany) or using microplate reader NOVOstar (BMG Labtech, Offenburg, Germany).
Detection of microorganism in human and mouse lung tissues
Gram-positive and gram-negative bacteria were detected using Gram Stain Kit (Abcam, Amsterdam, Netherlands). Briefly, after deparaffination and hydration, paraffin embedded lung tissues were incubated in Gentian violet solution and rinsed in water. Slides were incubated in Lugol’s iodine solution and rinsed with water. Slides were decolorized with Gram decolorizer and then incubated in carbol fuchsin. After washing, the tissue elements were counterstained with tartrazine solution.
Patch Clamping
CFBE cells were grown on coated glass cover slips. Patch pipettes were filled with a cytosolic-like solution containing (in mM) KCl 30, K-gluconate 95, NaH2PO4 1.2, Na2HPO4 4.8, EGTA 1, Ca -gluconate 0.758, MgCl2 1.03, D-glucose 5, ATP 3, pH 7.2. The intracellular (pipette) Ca2+ activity was 0.1 µM. The bath was perfused continuously with Ringer solution at a rate of 8 ml/min. Patch pipettes had an input resistance of 2–4 MW and whole cell currents were corrected for serial resistance. Currents were recorded using a patch clamp amplifier EPC9, and PULSE software (HEKA, Lambrecht, Germany) as well as Chart software (AD Instruments, Spechbach, Germany). The currents were corrected for the serial resistance. The acquisition frequency was 1 kHz. The signal was filtered by a low-pass Bessel filter. In regular intervals, the membrane voltage (Vc) was clamped from -100 to +100 mV in steps of 20 mV. If not voltage clamped, the cells were kept at their intrinsic membrane voltage in the current clamp mode. Membrane capacitance was measured using the PULSE software. Current density was calculated by dividing whole cell currents by cell capacitance.
Measurement of [Ca2+]i
Measurement of the intracellular Ca2+ concentration was performed as described previously [16]. In brief, after incubation with P. aeruginosa, cells were loaded with 2 μM Fura-2/AM and 0.02% Pluronic F-127 (Invitrogen, Darmstadt, Germany) to measure global changes in cytosolic Ca2+. Fluorescence was detected at 37°C, using an inverted microscope (Axiovert S100, Zeiss, Germany) and a high speed polychromator system (VisiChrome, Puchheim, Germany). Fura-2 was excited at 340/380 nm, and emission was recorded between 470 and 550 nm using a CCD-camera (CoolSnap HQ, Visitron Systems, Germany). After calibration intracellular Ca2+ concentrations were calculated.
Western Blotting
Protein was isolated from cells using a sample buffer containing 50 mM Tris-HCl, 150 mM NaCl, 50 mM Tris, 100 mM dithiothreitol, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1% protease inhibitor mixture (Sigma, Taufkirchen, Germany). Proteins were separated by 8.5 % SDS-PAGE and transferred to a polyvinyl membrane (GE Healthcare, Munich, Germany). Membranes were incubated with primary rabbit monoclonal [SP31] anti-TMEM16A antibody (#ab64085, Abcam, Cambridge, UK 1:500) overnight at 4°C. Proteins were visualized using horseradish peroxidase-conjugated secondary antibody and ECL detection kit (ECL™ Prime Western-Blot-System, Sigma-Aldrich, Taufkirchen, Germany) . GAPDH was used as a loading control.
Detection of IL-8, TNF-α, IL-1α and IL-1ß
IL-8, TNF-α, IL-1α and IL-1ß secretion was detected using quantikine colorimetric sandwich ELISA kits (R&D systems, Wiesbaden-Nordenstadt, Germany). After cells were infected with P. aeruginosa, cell culture supernates were collected. Particulates were removed by centrifugation and assay immediately according to the protocol for the company. The signal was detected using microplate reader NOVOstar (BMG Labtech, Offenburg, Germany).
Materials and statistical analysis
All compounds used were of highest available grade of purity. The TMEM16A-inhibitors and the other drugs used in the present study, such as ferrostain-1, coenzyme Q10, benzbromarone, niclosamide, Ani9, idebenone, CFTRinh172, and ST034307 were all from Sigma-Aldrich (Taufkirchen, Germany). Data are reported as mean ± SEM. Student’s t test for unpaired samples and ANOVA were used for statistical analysis. P < 0.05 was accepted as significant difference. Data are expressed as mean ± SEM. Differences among groups were analyzed using one-way ANOVA, followed by a Bonferroni test for multiple comparisons. An unpaired t test was applied to compare the differences between two groups.
Results
Bacterial colonization and lipid peroxidation in CF airways
In the present study various aspect were examined in lung tissue sections from in total 5 CF- and 5 non-CF patients. Gram-negative and Gram-positive bacteria were detected in lung tissue slices of 3 CF patients, while no bacteria were detected in lung slices of 3 non-CF (asthmatic) patients (Fig. 1A). Lipid peroxidation was analyzed in the airway epithelium using 4-Hydroxy-2-nonenal (4HNE) staining. Immunofluorescence signals from 4HNE stainings in the airway epithelium were determined by marking the regions of interest (ROI; epithelium only). A large number of images (n=79-291) were analyzed from each patient lung. Dark precipitations indicated membrane lipid peroxidation in 5 CF patients, while no clear lipid peroxidation could be detected in 3 non-CF airways (Fig. 1B, C). These data confirm earlier findings and suggest a contribution of bacterial infections to lipid peroxidation in human airway epithelial cells, as suggested by Dar et al. [6].
Role of CFTR for lipid peroxidation
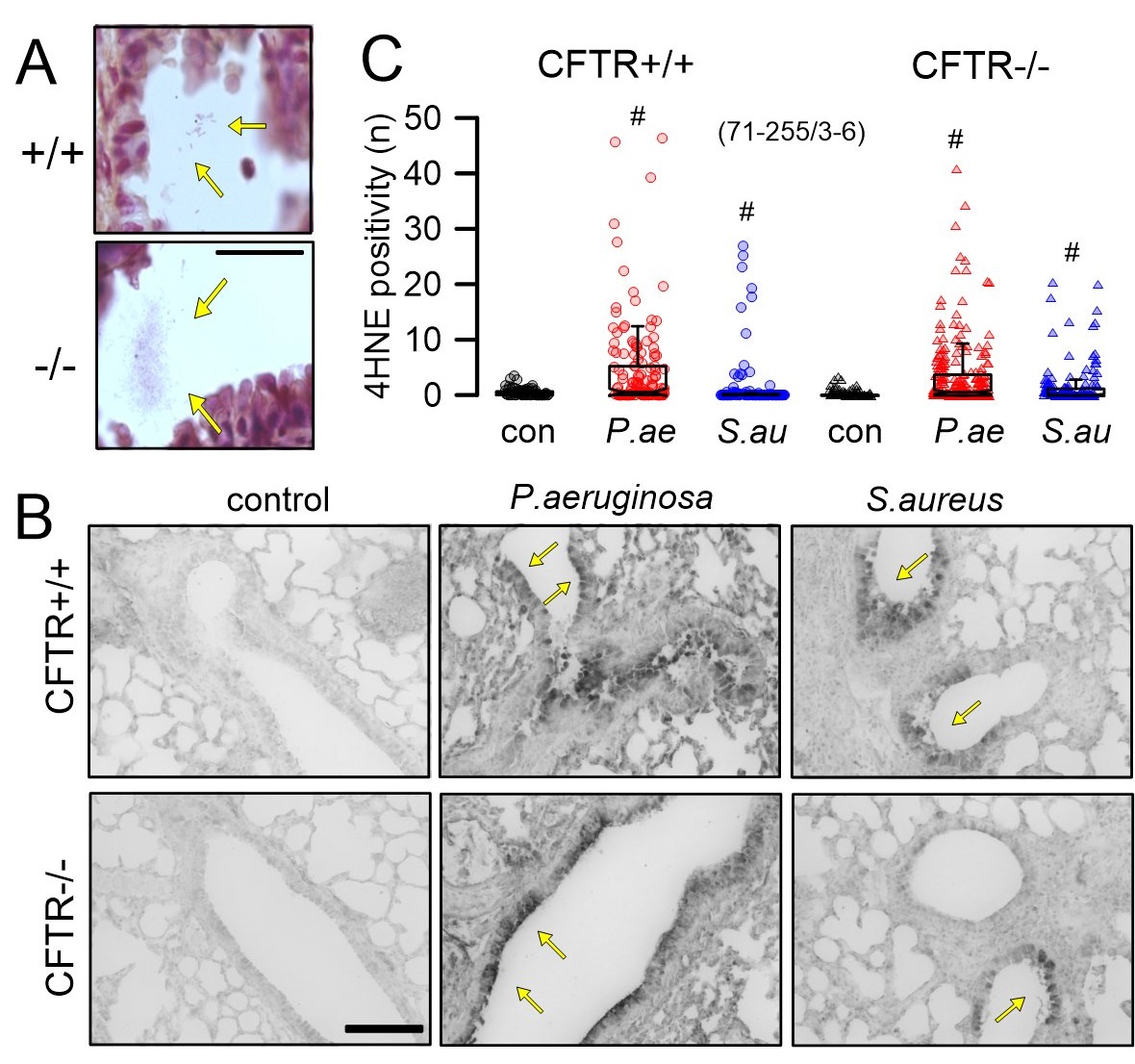
The role of bacterial infection for lipid peroxidation was further analyzed in mouse lungs, in order to determine whether lipid peroxidation is correlated with bacterial infection. Moreover, it was essential to determine examine differences regarding spontaneous lipid peroxidation (in the absence of bacteria) between CFTR+/+ and CFTR-/- mice. To that end, mice were infected with P. aeruginosa or S. aureus as described earlier [15]. After intratracheal infection, bacteria could be detected in lungs of both CFTR+/+ and CFTR-/- mice (Fig. 2A). Lipid peroxidation was detected in airways of mice infected with P. aeruginosa or S. aureus, but not in non-infected control animals. Thus, 4HNE staining showed no signals in lungs from CFTR+/+ and CFTR-/- control animals, but showed lipid peroxidation in P. aeruginosa infected animals and, to a weaker extend, also in S. aureus infected animals (Fig. 2B, C). The data suggest i) oxidative stress and possible ferroptosis only after bacterial infection, which ii) is not specific to P. aeruginosa, and iii) not dependent on expression of CFTR. It is well known that CFTR is expressed at only low levels in mouse airways, which demonstrate only little cAMP-activated Cl- secretion [17, 18]. In contrast to CFTR-knockout mice, which demonstrate an only weak CF-phenotype, knockout of CFTR in piglets induced a full-blown CF disease [19]. We therefore also analyzed lipid peroxidation in airways of newborn piglets lacking expression of CFTR [14, 20]. Interestingly, 4HNE-staining suggested lipid peroxidation in lungs from CFTR-/- but not from CFTR+/+ animals, suggesting that lack of pulmonary CFTR-expression predisposes to oxidative stress even in the absence of bacterial infection (Supplementary Fig. 1 – for all supplementary material see www.cellphysiolbiochem.com). Similar has also been observed in young infants [21].
P. aeruginosa induced ROS and lipid peroxidation in human airway epithelial cells
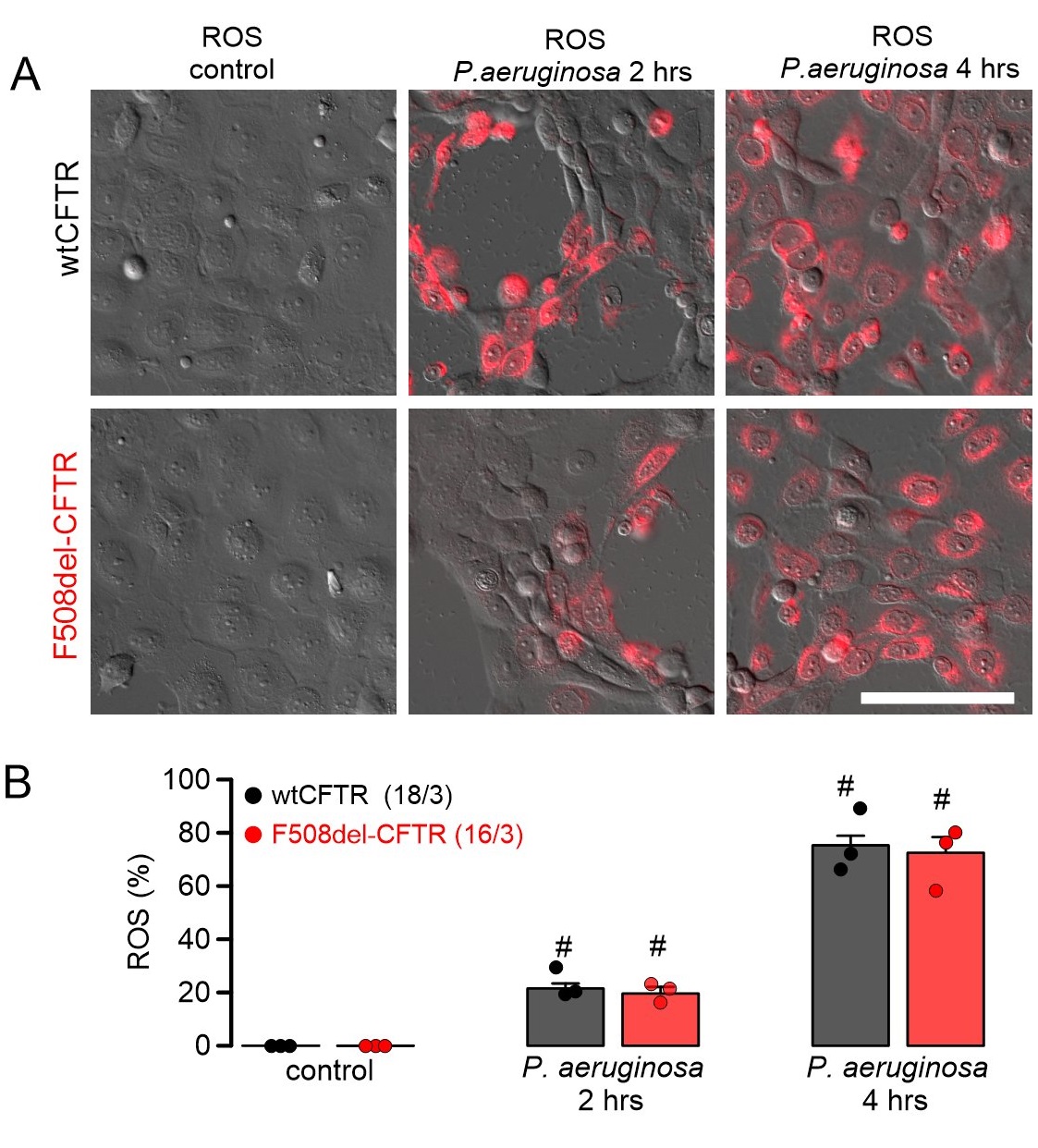
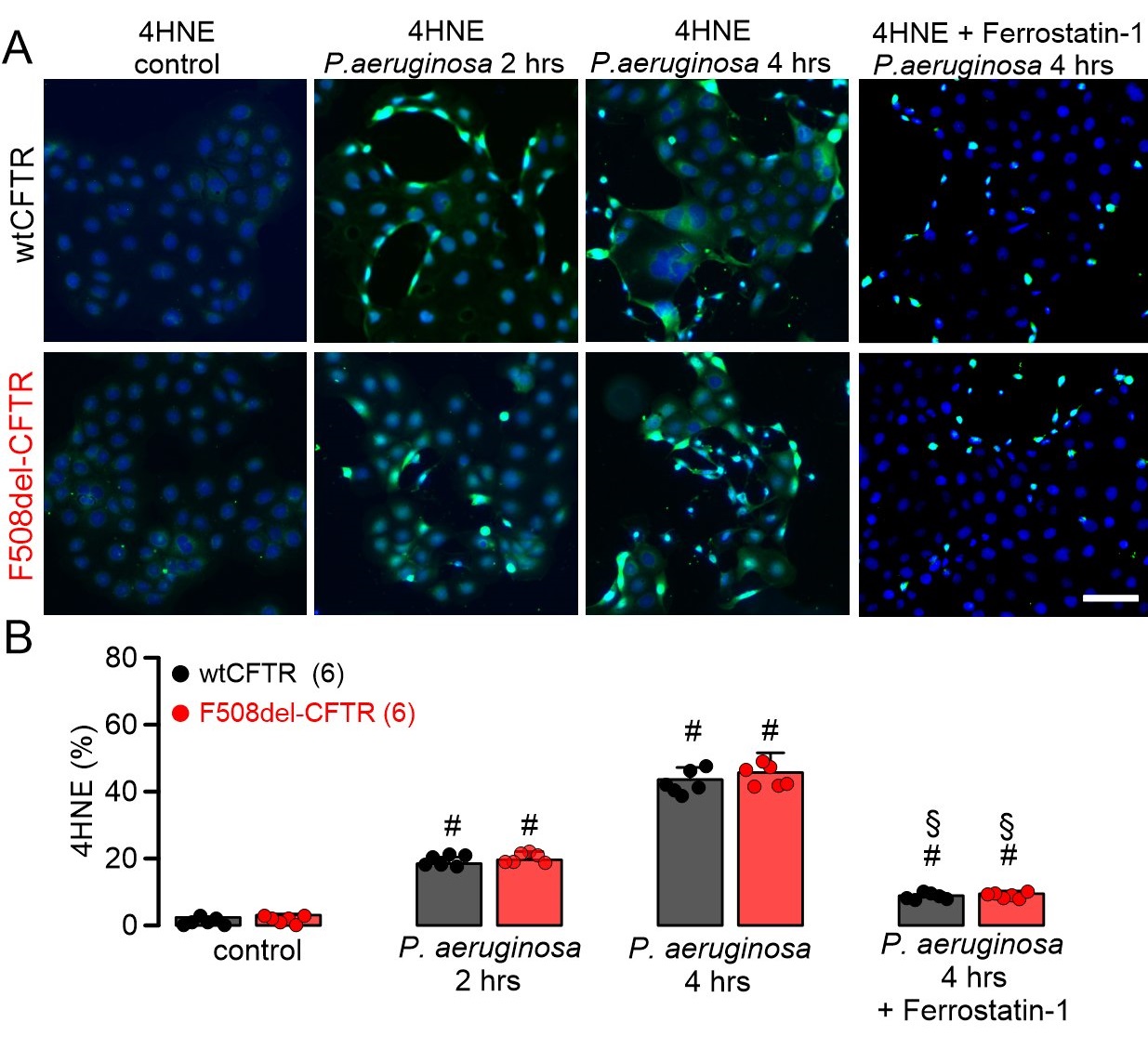
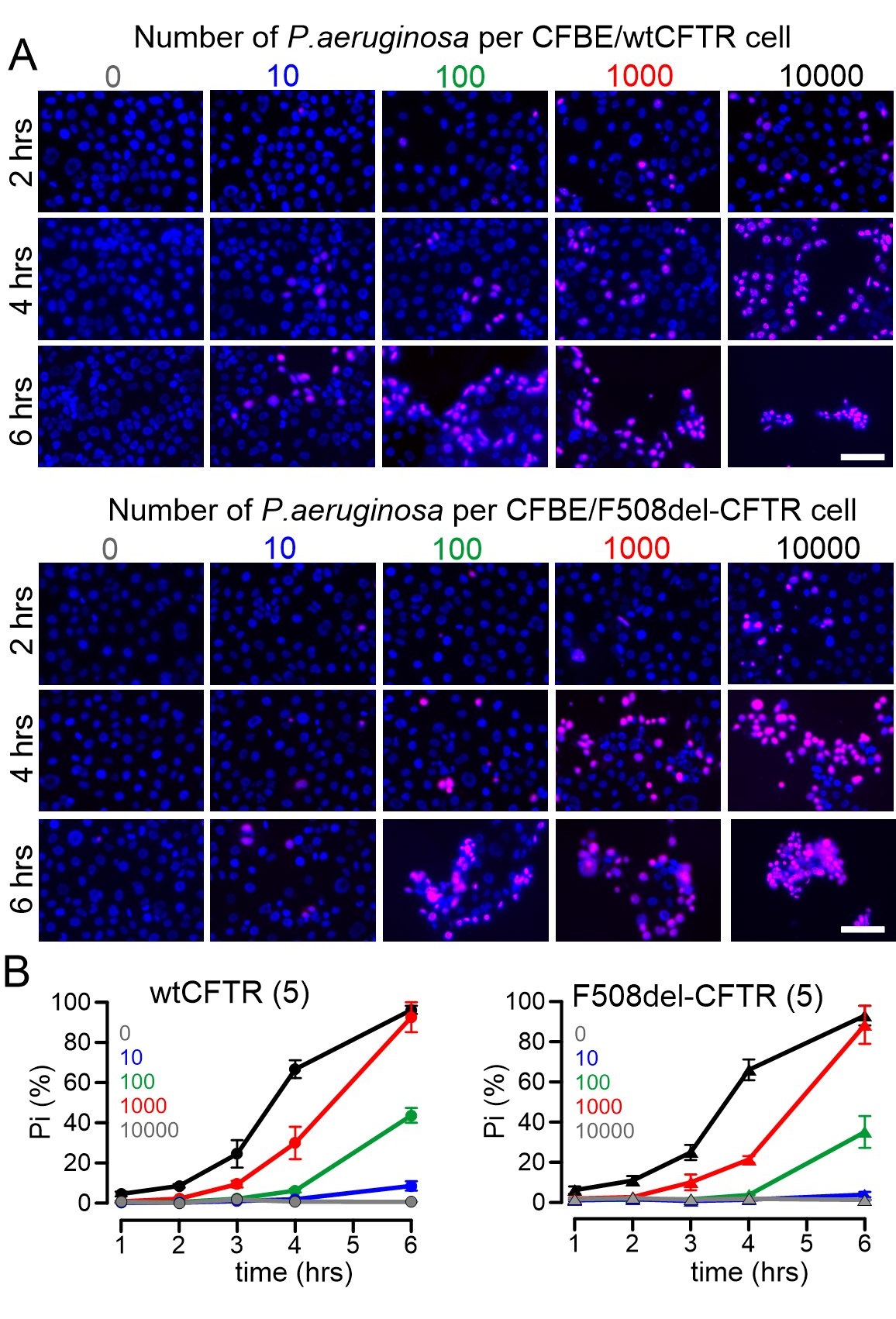
ROS formation was analyzed in CFBE human airway epithelial cells stably expressing wtCFTR or F508del-CFTR. ROS was not detected under control conditions, but was clearly induced when cells were exposed to P. aeruginosa (Fig. 3). Moreover, lipid peroxidation induced by tert-butyl hydroperoxide was not different between CFBE/wtCFTR and CF/F508del-CFTR cells, and was detected in 82.2 ± 9.2 % (n=3; wtCFTR) and 87.6 ± 8.8 % (n=3; F508del-CFTR), respectively, of all cells. These data suggest that ROS-buffer capacity is similar in airway epithelial cells expressing wtCFTR or F508del-CFTR. Exposure to P. aeruginosa and induction of ROS was paralleled by lipid peroxidation, which could be clearly inhibited by ferrostatin-1, suggesting induction of ferroptosis by P. aeruginosa (Fig. 4). Moreover, exposure to P. aeruginosa and induction of ferroptosis induced cell death as detected by propidium iodide permeabilization. Again, the rate of cell death was equal for both cells expressing wtCFTR or F508del-CFTR (Fig. 5).
TMEM16A contributes to cell death induced by P. aeruginosa
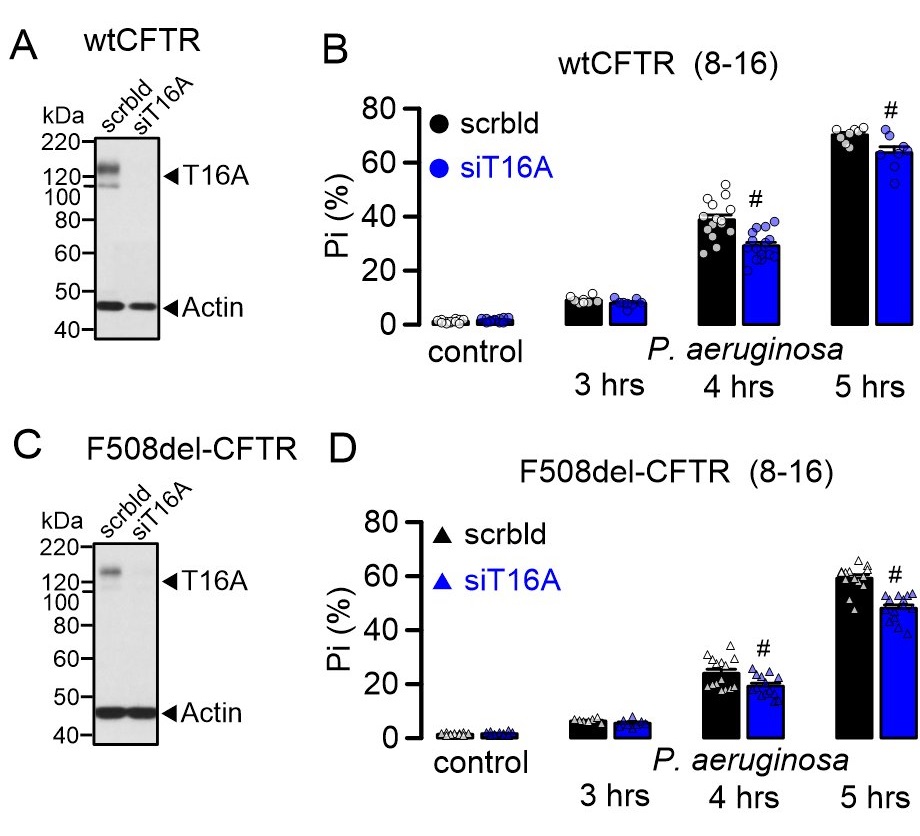
Our previous studies pointed out to a contribution of TMEM16A and TMEM16F to regulated cell death in airways [8] and polycystic kidney disease, respectively [12]. We therefore asked whether TMEM16A also contributes to ferroptotic cell death induced by P. aeruginosa. To that end, TMEM16A was knocked down by siRNA (siT16A) in CFBE cells (Fig. 6A, C). siRNAs were transfected using standard protocols for electroporation or, alternatively, lipofectamine 3000 was transfected as control. All cells transfected with siRNA were used for experiments 48-72 h after transfection. Successful knockdown was verified by Western-blotting, which indicated a complete knockdown for both ANO1 and ANO6. Cell death was determined 1-5 hours after applying P. aeruginosa to the cells. Indeed, cell death induced by 1000 P. aeruginosa per cell was partially inhibited by knockdown of TMEM16A in CFBE/wtCFTR and CFBE/F508del-CFTR cells (Fig. 6B, D). In contrast, knockdown of TMEM16F did not inhibit P. aeruginosa induced cell death (Supplementary Fig. 2). However, the missing effect of siTMEM16F on P. aeruginosa-induced cell death might also be due to the very low expression levels of TMEM16F in CFBE cells, which could not be detected by Western blotting. Expression of TMEM16F is somewhat higher in Calu3 airway epithelial cells [22] and knockdown of TMEM16F showed some inhibitory effect on P. aeruginosa-induced cell death (Supplementary Fig. 3).
Inhibition of P. aeruginosa-induced cell death by inhibitors of ROS and ferroptosis, but not by inhibitors of TMEM16A
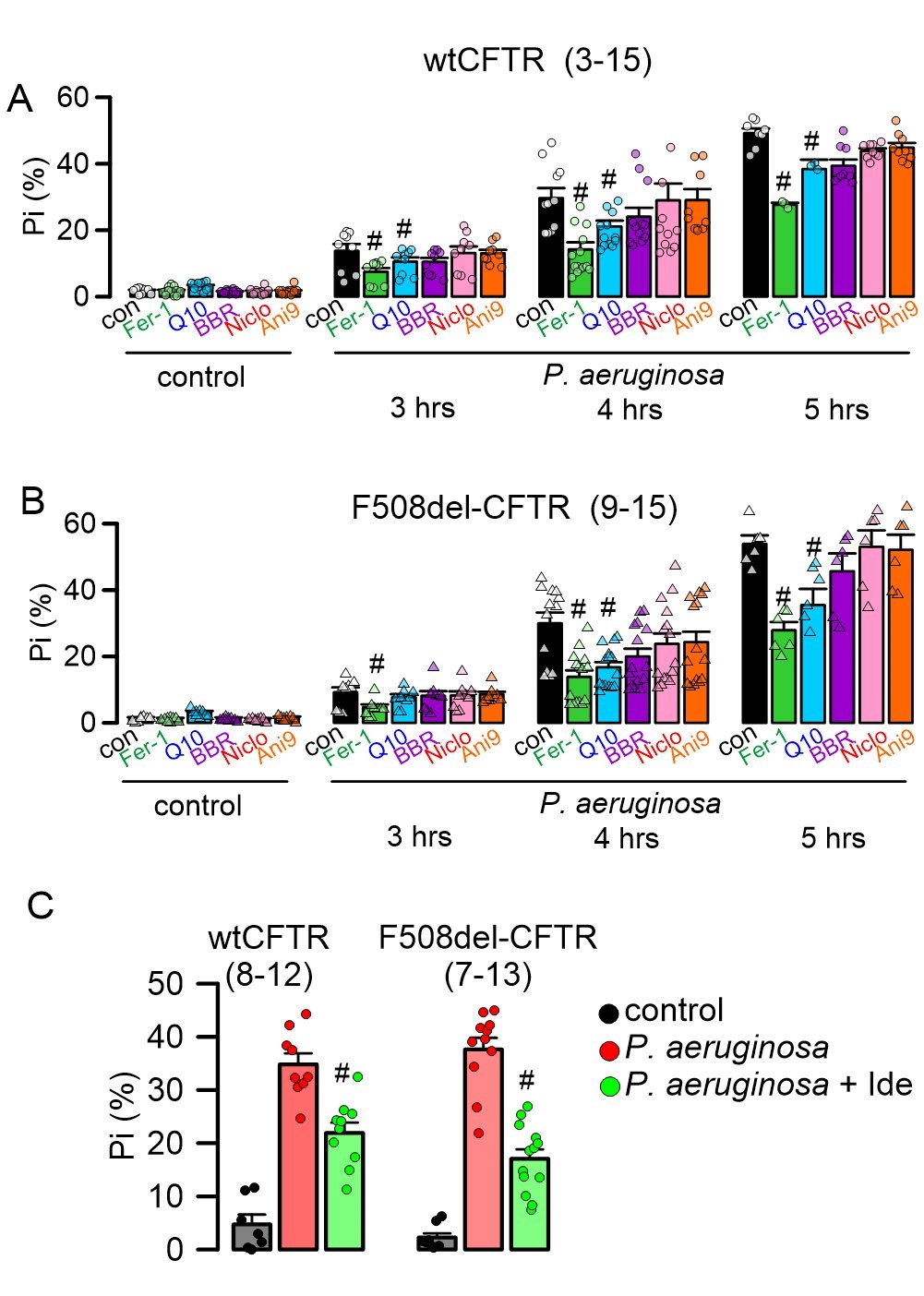
Because knockdown of TMEM16 proteins partially inhibited P. aeruginosa induced cell death, we examined whether inhibitors of TMEM16A or TMEM16F, (benzbromarone, niclosamide, Ani9) and antioxidants/ferroptosis-inhibitors (coenzyme Q10, ferrostatin-1) interfere with cell death in CFBE/wtCFTR and CFBE/F508del-CFTR cells. Coenzyme Q10 and ferrostatin-1 inhibited P. aeruginosa-induced cell death significantly, while the TMEM16A-inhibitors were without any significant effects (Fig. 7A, B). In addition, we examined the effect of another ROS scavenger, idebenone, which is also known to inhibit TMEM16A (Fig. 7C). Idebenone is a FDA-approved drug. We found that it reduces cell death induced by P. aeruginosa significantly (Fig. 7C). Benzbromarone, niclosamide, and idebenone are all FDA-approved drugs for the treatment of hyperuricemia, tape-worm infection and eye disease, respectively, while Q10 is an antioxidant used as a food supplement. Idebenone has been shown to inhibit TMEM16A and to operate as an antioxidant [11, 12, 23]. The data suggest antioxidants and inhibition of ferroptosis as an effective therapy to reduce P. aeruginosa induced cell death.
P. aeruginosa increases basal intracellular Ca2+ concentrations and activates CFTR
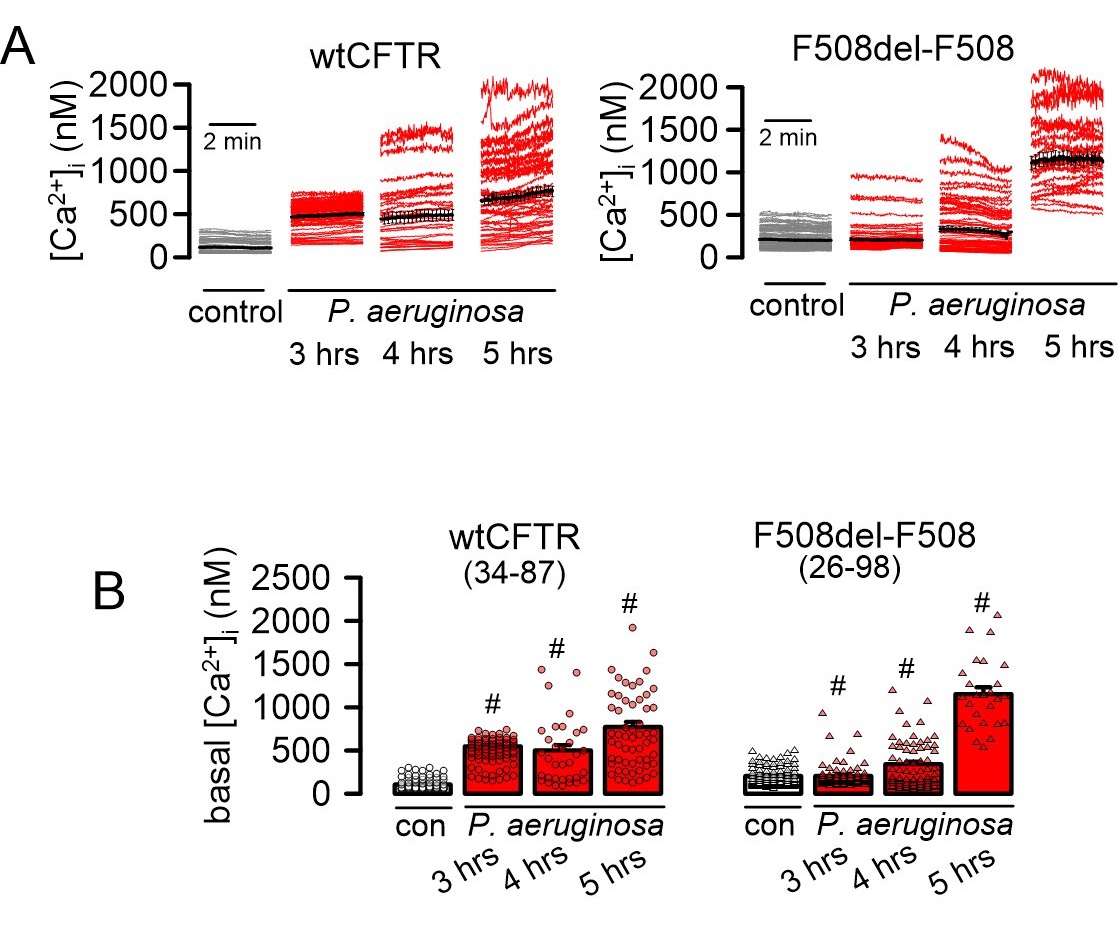
Because ROS formation is known to affect cytosolic Ca2+, we examined whether exposure to P. aeruginosa induces an increase in the intracellular Ca2+ concentration. Indeed, continuous exposure to P. aeruginosa for up to 5 hours gradually increased basal Ca2+ levels in both CFBE/wtCFTR and CFBE/F508del-CFTR cells (Fig. 8A, B). In patch clamp recordings we detected enhanced membrane currents in CFBE/wtCFTR cells exposed to P. aeruginosa, which was not observed in CFBE cells expressing F508del-CFTR. Removal of extracellular Cl- strongly inhibited membrane conductance and shifted the reversal potential in CFBE/wtCFTR cells, indicating activation of a whole cell Cl- current. Similarly, the CFTR-inhibitor 172 also blocked the P. aeruginosa-induced current, indicating activation of CFTR Cl- currents, which are absent in F508del-CFTR expressing cells (Fig. 9A-F). Based on previous observations, we speculated that increase in basal Ca2+ by P. aeruginosa activates CFTR by stimulation of a Ca2+ dependent adenylate cyclase, possibly ADCY1. In fact, the ADCY1-inhibitor ST034307 also inhibited activated of CFTR, suggesting a defense mechanism in non-CF airways, which is absent in airways of people with CF (Fig. 9G-I).
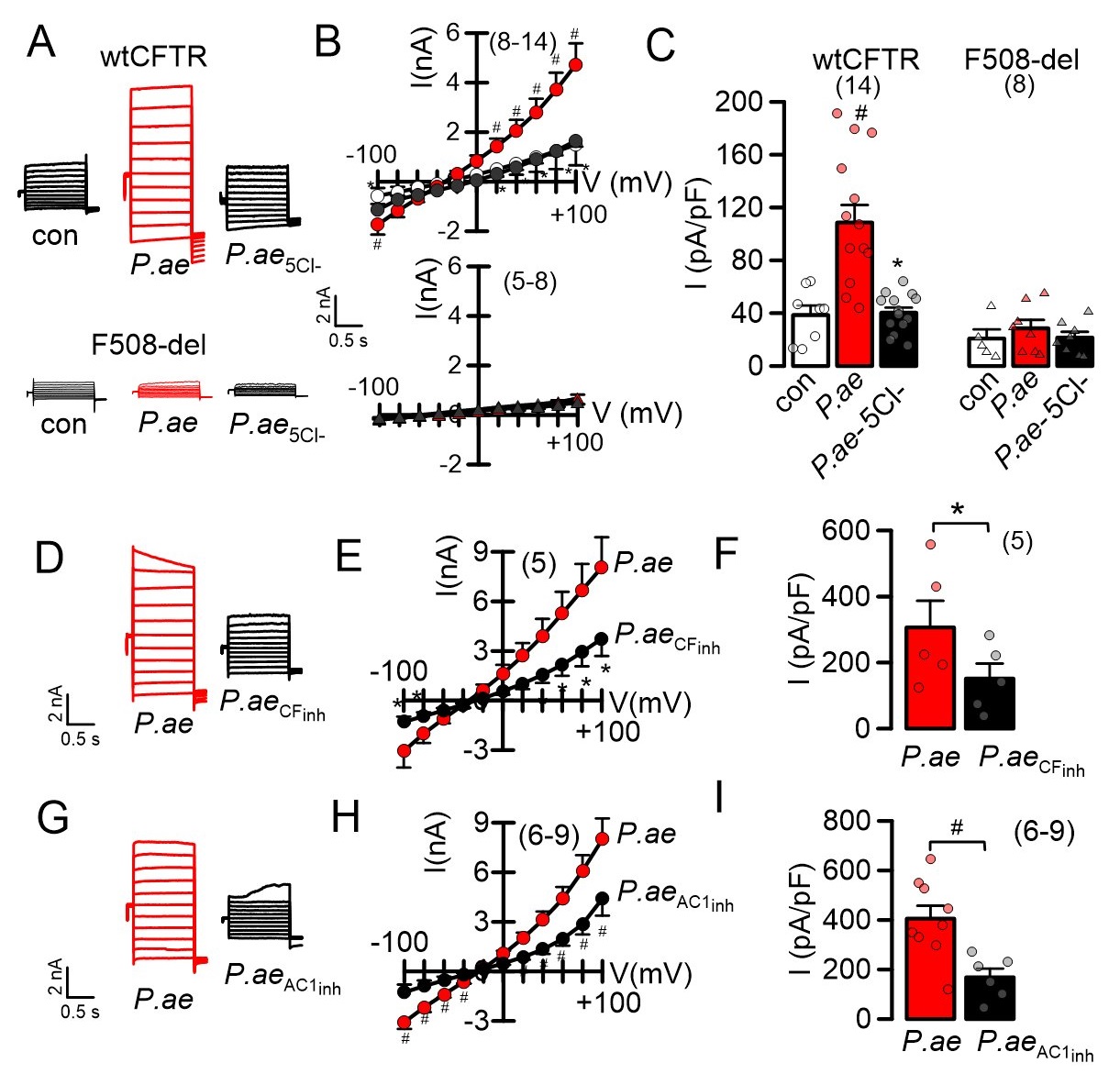
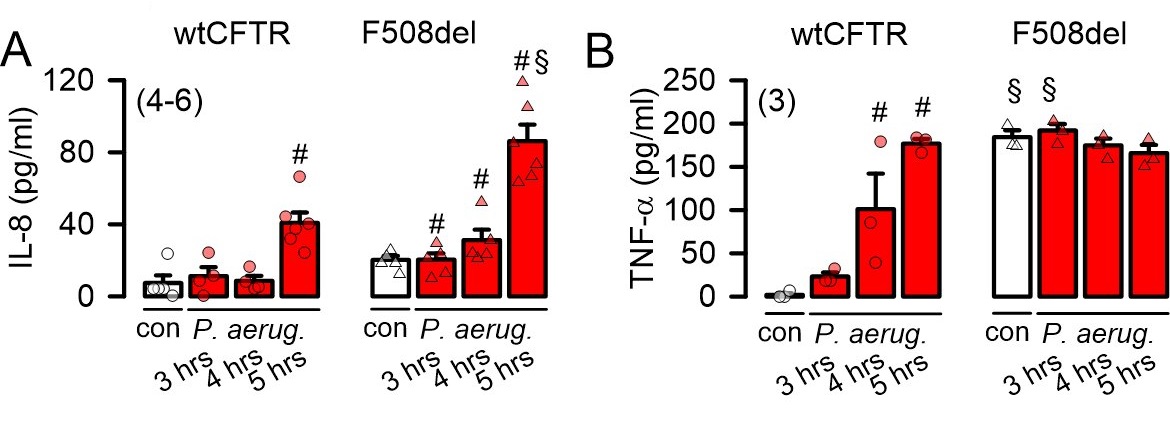
P. aeruginosa cause release of cytokines
IL-8 produced in the bronchial epithelium improves lung immunity towards bacterial infection, but also drives lung damage through persistent inflammation, causing lung remodelling [24]. We examined the release of four cytokines, IL-8, TNF-α, IL-1α and IL-1ß by exposure of CFBE/wtCFTR or CFBE/F508del-CFTR cells to P. aeruginosa. P. aeruginosa induced release of IL-8 and TNF-α in both wtCFTR and F508del-CFTR expressing cells, which was augmented in cells expressing F508del-CFTR (Fig. 10A). IL-1α and IL-1ß were not released upon exposure to P. aeruginosa (Table 1). Notably, release of TNF-α by CFBE/F508del-CFTR cells was high even in the absence of P. aeruginosa, strongly suggesting a proinflammatory phenotype in human airways expressing F508del-CFTR [25, 26]. Taken together, the present results suggest a hypoxic predisposition and lipid peroxidation in CF airways, which is augmented by infection with P. aeruginosa. The results provide evidence for a contribution of TMEM16A/F to ferroptotic cell damage of airway epithelial cells, which can be inhibited by antioxidants and inhibitors of ferroptosis.
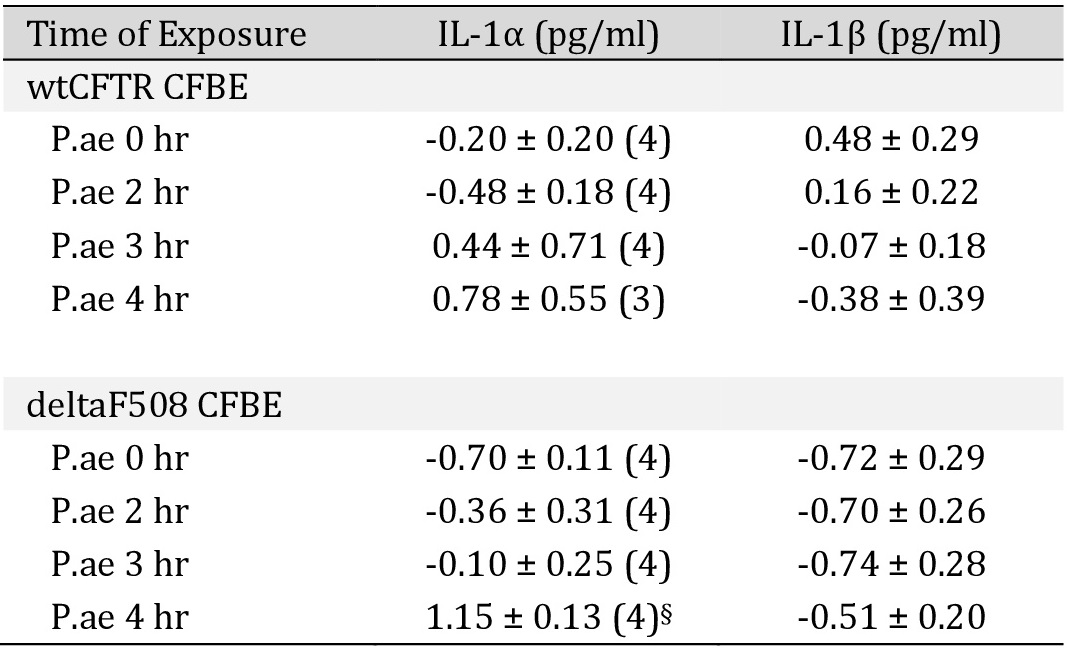
Release of IL-1α and IL-1β induced by P. aeruginosa in CFBE cells
Discussion
The present data and earlier reports suggest enhanced lipid peroxidation in the airways of CF patients. In CFTR-knockout piglets, lipid peroxidation was observed even in the absence of P. aeruginosa. This was not seen in airways of CFTR-knockout mice. In contrast to mice, CF-piglets develop a severe lung disease [27]. Similar to humans, also in piglets airway function largely relies on CFTR expression, unlike in mouse airways which express much lower levels of CFTR. It is important to note that structural changes were observed in lungs of infants with CF and in newborn piglets lacking expression of CFTR, such as bronchiectasis, altered orientation of smooth muscle bundles and sometimes an irregular epithelium [14, 20, 28, 29]. These structural and developmental changes may also contribute to hypoxic regions within the CF lung, with higher local ROS levels leading to lipid peroxidation.
Apart from these structural predispositions, infection with P. aeruginosa clearly induced comparable lipid peroxidation in both wt and CFTR-knockout mice, and this was reproduced in isolated human airway epithelial cells expressing wtCFTR or F508del-CFTR. The recently identified novel modifier gene TAS2R38 that was identified in patients with cystic fibrosis, may have a significant impact on chronic lung colonization by P. aeruginosa [30, 31]. Lipid peroxidation induced ferroptotic cell death that could be antagonized by antioxidants and ferrostatin-1. Although siRNA-knockdown of TMEM16A and TMEM16F suggest a certain role of these proteins in cell death, none of the TMEM16 inhibitors attenuated cells death, unlike in previous studies [8, 32, 33]. In renal epithelial cells TMEM16A is activated by lipid peroxidation and thereby contributes to renal cyst formation [12]. The present data show that P. aeruginosa induced increase in intracellular Ca2+ that does not activate TMEM16A currents, as demonstrated by the fact that no currents were activated in F508del-expressing cells, which, however, express TMEM16A. Thus inhibitors of the TMEM16A/F currents such as niclosamide, benzbromarone, and Ani9 will not affect P. aeruginosa induced cell death. TMEM16A/F most likely support ferroptotic cell death by enhancing local intracellular Ca2+ levels, as described earlier [34].
The relationship between oxidative stress airway ion transport and cell death is well recognized [35]. P. aeruginosa enhanced basal Ca2+ and activated CFTR in wtCFTR-expressing cells, which has also been observed in earlier studies [36, 37]. Schwarzer et al. reported P. aeruginosa homoserine lactone that activates store-operated cAMP [38]. These results are supported by the present data, indicating Ca2+ activated adenylate cyclase as the source for Ca2+-induced cAMP with subsequent activation of CFTR. The results correlate well with the previous finding of hydrogen peroxide stimulation of CFTR due to Epac-mediated activation of soluble adenylate cyclase [39]. Thus, in the healthy lung, bacterial components activate electrolyte secretion to augment mucociliary clearance, a mechanism that does not operate in the absence of functional CFTR.
The present data strongly suggest the use of antioxidants in cystic fibrosis [40]. Idebenone is a potent antioxidant compound and inhibitor of TMEM16A [11, 12, 23]. Idebenone is approved for the use in Duchenne muscular dystrophy and Leber hereditary optic neuropathy [41], and may also improve lipid peroxidation and cell death in cystic fibrosis lungs. Moreover, direct inhibition of P. aeruginosa-induced cell death in CF lungs and other forms of bacterial pneumonia, by new generations of ferroptosis inhibitors may significantly improve lung inflammation [42].
We thank Dr. Ines Cabrita (Physiologisches Institut, Universität Regensburg) for performing Ca2+ measurements. The lung tissue from piglets was generously provided by Prof. Dr. Klymiuk (Institute of Molecular Animal Breeding and Biotechnology, Gene Center, Ludwig-Maximilians-Universität München). Wild type (CFTR+/+) and CFTR knockout (CFTR-/-) piglet were generated in laboratory of Prof. Dr. N. Klymiuk.
All original data are available on request.
Author Contributions
Conceptualization, J.O., R.S., and K.K.; Methodology, J.O., R.S., and E.G.; formal analysis, J.O., R.S., and K.K.; Investigation, J.O., R.S., and K.K.; Data curation, J.O., R.S., and K.K.; Writing—original draft preparation, J.O., R.S., and K.K.; Writing—review and editing, J.O., R.S., E.G., M.K., and K.K..
Funding
Supported by DFG project number 387509280, SFB 1350 (project A3), DFG KU756/14-1
and Mukoviszidose e.V..
Statement of Ethics
Analysis of human lung sections has been approved by the Ethikkommission des Universitätsklinikum Essen (University of Duisburg-Essen, Hufelandstrasse 55, 45122 Essen, Germany), Genehmigungsnummer 17-7326-BO. The Experimental work on wt and CFTR-/-
mice was approved by Bezirksregierung Duesseldorf, Duesseldorf, Germany. Permission numbers G1380-13, G1376-13, G1121-10, AZ 84.02.04.2015.A064.
All work on CF pigs has been conducted under the supervision of the responsible regulatory authority (Regierung von Oberbayern) and animals were sacrificed for scientific purposes according to the German Animal Welfare law §4.
The authors declare that no conflict of interests exists.
| 1 Trudel S, Kelly M, Fritsch J, Nguyen-Khoa T, Therond P, Couturier M, Dadlez M, Debski J, Touqui L, Vallee B, Ollero M, Edelman A, Brouillard F: Peroxiredoxin 6 fails to limit phospholipid peroxidation in lung from Cftr-knockout mice subjected to oxidative challenge. PLoS One 2009;4:e6075. https://doi.org/10.1371/journal.pone.0006075 |
||||
| 2 Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, Oliveira-Munding CC, Van Heeckeren AM, Barr ML, von Kurthy G, Schmid KW, Weller M, Tummler B, Lang F, Grassme H, Doring G, Gulbins E: Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med 2008;14:382-391. https://doi.org/10.1038/nm1748 |
||||
| 3 Hull J, Vervaart P, Grimwood K, Phelan P: Pulmonary oxidative stress response in young children with cystic fibrosis. Thorax 1997;52:557-560. https://doi.org/10.1136/thx.52.6.557 |
||||
| 4 Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, Massi-Benedetti C, Borghi M, Puccetti M, Lucidi V, Colombo C, Fiscarelli E, Lass-Florl C, Majo F, Cariani L, Russo M, Porcaro L, Ricciotti G, Ellemunter H, Ratclif L, et al.: IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun 2016;7:10791. https://doi.org/10.1038/ncomms10791 |
||||
| 5 Huus KE, Joseph J, Zhang L, Wong A, Aaron SD, Mah TF, Sad S: Clinical Isolates of Pseudomonas aeruginosa from Chronically Infected Cystic Fibrosis Patients Fail To Activate the Inflammasome during Both Stable Infection and Pulmonary Exacerbation. J Immunol 2016;196:3097-3108. https://doi.org/10.4049/jimmunol.1501642 |
||||
| 6 Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting HC, Tyurin VA, Krieger J, St Croix CM, Watkins S, Bayir E, Mao G, Ambruster C, Kapralov A, Wang H, Parsek MH, Anthonymuthu TS, Ogunsola AF, Flitter BA, Freedman CJ, Gaston JR, et al.: Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest 2018;128:4639-4653. https://doi.org/10.1172/JCI99490 |
||||
| 7 Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE: CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J 2003;22:1981-1989. https://doi.org/10.1093/emboj/cdg194 |
||||
| 8 Simoes F, Ousingsawat J, Wanitchakool P, Fonseca A, Cabrita I, Benedetto R, Schreiber R, Kunzelmann K: CFTR supports cell death through ROS-dependent activation of TMEM16F (anoctamin 6). Pflugers Arch 2018;470:305-314. https://doi.org/10.1007/s00424-017-2065-0 |
||||
| 9 Ousingsawat J, Schreiber R, Kunzelmann K: TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers (Basel) 2019;11:pii:E625. https://doi.org/10.3390/cancers11050625 |
||||
| 10 Martins JR, Faria D, Kongsuphol P, Reisch B, Schreiber R, Kunzelmann K: Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc Natl Acad Sci U S A 2011;108:18168-18172. https://doi.org/10.1073/pnas.1108094108 |
||||
| 11 Schreiber R, Ousingsawat J, Wanitchakool P, Sirianant L, Benedetto R, Reiss K, Kunzelmann K: Regulation of TMEM16A/ANO1 and TMEM16F/ANO6 ion currents and phospholipid scrambling by Ca2+ and plasma membrane lipid J Physiology (London) 2018;596:217-229. https://doi.org/10.1113/JP275175 |
||||
| 12 Schreiber R, Buchholz B, Kraus A, Schley G, Scholz J, Ousingsawat J, Kunzelmann K: Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J Am Soc Nephrol 2019;30:228-242. https://doi.org/10.1681/ASN.2018010039 |
||||
| 13 Montgomery ST, Mall MA, Kicic A, Stick SM: Hypoxia and sterile inflammation in cystic fibrosis airways: mechanisms and potential therapies. Eur Respir J 2017;49:1600903. https://doi.org/10.1183/13993003.00903-2016 |
||||
| 14 Klymiuk N, Mundhenk L, Kraehe K, Wuensch A, Plog S, Emrich D, Langenmayer MC, Stehr M, Holzinger A, Kroner C, Richter A, Kessler B, Kurome M, Eddicks M, Nagashima H, Heinritzi K, Gruber AD, Wolf E: Sequential targeting of CFTR by BAC vectors generates a novel pig model of cystic fibrosis. J Mol Med (Berl) 2012;90:597-608. https://doi.org/10.1007/s00109-011-0839-y |
||||
| 15 Grassme H, Jendrossek V, Riehle A, von Kurthy G, Berger J, Schwarz H, Weller M, Kolesnick R, Gulbins E: Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat Med 2003;9:322-330. https://doi.org/10.1038/nm823 |
||||
| 16 Ousingsawat J, Cabrita I, Wanitchakool P, Sirianant L, Krautwald S, Linkermann A, Schreiber R, Kunzelmann K: Ca2+ signals, cell membrane disintegration, and activation of TMEM16F during necroptosis. Cell Mol Life Sci 2016;74:173-181. https://doi.org/10.1007/s00018-016-2338-3 |
||||
| 17 Rochelle LG, Li DC, Ye H, Lee E, Talbot CR, Boucher RC: Distribution of ion transport mRNAs throughout murine nose and lung. Am J Physiol 2000;279:L14-L24. https://doi.org/10.1152/ajplung.2000.279.1.L14 |
||||
| 18 Lee RJ, Foskett JK: Why mouse airway submucosal gland serous cells do not secrete fluid in response to cAMP stimulation. J Biol Chem 2012;287:38316-38326. https://doi.org/10.1074/jbc.M112.412817 |
||||
| 19 Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, et al.: Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2010;2:29ra31. https://doi.org/10.1126/scitranslmed.3000928 |
||||
| 20 Benedetto R, Centeio R, Ousingsawat J, Schreiber R, Janda M, Kunzelmann K: Transport properties in CFTR-/- knockout piglets suggest normal airway surface liquid pH and enhanced amiloride-sensitive Na(+) absorption. Pflugers Arch 2020;472:1507-1519. https://doi.org/10.1007/s00424-020-02440-y |
||||
| 21 Kettle AJ, Turner R, Gangell CL, Harwood DT, Khalilova IS, Chapman AL, Winterbourn CC, Sly PD: Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur Respir J 2014;44:122-129. https://doi.org/10.1183/09031936.00170213 |
||||
| 22 Benedetto R, Ousingsawat J, Cabrita I, Pinto M, Lerias J, Wanitchakool P, Schreiber R, Kunzelmann K: Plasma membrane localized TMEM16 Proteins are Indispensable for expression of CFTR. J Mol Med 2019;97:711-722. https://doi.org/10.1007/s00109-019-01770-4 |
||||
| 23 Seo Y, Park J, Kim M, Lee HK, Kim JH, Jeong JH, Namkung W: Inhibition of ANO1/TMEM16A Chloride Channel by Idebenone and Its Cytotoxicity to Cancer Cell Lines. PLoS ONE 2015;10:e0133656. https://doi.org/10.1371/journal.pone.0133656 |
||||
| 24 Reynolds CJ, Quigley K, Cheng X, Suresh A, Tahir S, Ahmed-Jushuf F, Nawab K, Choy K, Walker SA, Mathie SA, Sim M, Stowell J, Manji J, Pollard T, Altmann DM, Boyton RJ: Lung Defense through IL-8 Carries a Cost of Chronic Lung Remodeling and Impaired Function. Am J Respir Cell Mol Biol 2018;59:557-571. https://doi.org/10.1165/rcmb.2018-0007OC |
||||
| 25 Martins JR, Kongsuphol P, Sammels E, Daimène S, AlDehni F, Clarke L, Schreiber R, DeSmedt H, Amaral MD, Kunzelmann K: F508del-CFTR increases intracellular Ca2+ signaling that causes enhanced calcium-dependent Cl- conductance in cystic fibrosis. Biochim Biophys Acta 2011;1812:1385-1392. https://doi.org/10.1016/j.bbadis.2011.08.008 |
||||
| 26 Sutanto EN, Kicic A, Foo CJ, Stevens PT, Mullane D, Knight DA, Stick SM: Innate inflammatory responses of pediatric cystic fibrosis airway epithelial cells: effects of nonviral and viral stimulation. Am J Respir Cell Mol Biol 2011;44:761-767. https://doi.org/10.1165/rcmb.2010-0368OC |
||||
| 27 Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, Stoltz DA, Welsh MJ: Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014;345:818-822. https://doi.org/10.1126/science.1255825 |
||||
| 28 Long FR, Williams RS, Castile RG: Structural airway abnormalities in infants and young children with cystic fibrosis. J Pediatr 2004;144:154-161. https://doi.org/10.1016/j.jpeds.2003.09.026 |
||||
| 29 Mott LS, Graniel KG, Park J, de Klerk NH, Sly PD, Murray CP, Tiddens H, Stick SM: Assessment of early bronchiectasis in young children with cystic fibrosis is dependent on lung volume. Chest 2013;144:1193-1198. https://doi.org/10.1378/chest.12-2589 |
||||
| 30 Castaldo A, Cernera G, Iacotucci P, Cimbalo C, Gelzo M, Comegna M, Di Lullo AM, Tosco A, Carnovale V, Raia V, Amato F: TAS2R38 is a novel modifier gene in patients with cystic fibrosis. Sci Rep 2020;10:5806. https://doi.org/10.1038/s41598-020-62747-9 |
||||
| 31 Adappa ND, Workman AD, Hadjiliadis D, Dorgan DJ, Frame D, Brooks S, Doghramji L, Palmer JN, Mansfield C, Reed DR, Cohen NA: T2R38 genotype is correlated with sinonasal quality of life in homozygous ΔF508 cystic fibrosis patients. Int Forum Allergy Rhinol 2016;6:356-361. https://doi.org/10.1002/alr.21675 |
||||
| 32 Kunzelmann K, Ousingsawat J, Benedetto R, Cabrita I, Schreiber R: Contribution of Anoctamins to Cell Survival and Cell Death. Cancers (Basel) 2019;19:E382. https://doi.org/10.3390/cancers11030382 |
||||
| 33 Zeng JW, Chen BY, Lv XF, Sun L, Zeng XL, Zheng HQ, Du YH, Wang GL, Ma MM, Guan YY: Transmembrane member 16A participates in hydrogen peroxide-induced apoptosis by facilitating mitochondria-dependent pathway in vascular smooth muscle cells. Br J Pharmacol 2018;175:3669-3684. https://doi.org/10.1111/bph.14432 |
||||
| 34 Cabrita I, Benedetto R, Fonseca A, Wanitchakool P, Sirianant L, Skryabin BV, Schenk LK, Pavenstadt H, Schreiber R, Kunzelmann K: Differential effects of anoctamins on intracellular calcium signals. FASEB J 2017;31:2123-2134. https://doi.org/10.1096/fj.201600797RR |
||||
| 35 O'Grady SM: Oxidative stress, autophagy and airway ion transport. Am J Physiol Cell Physiol 2019;316:C16-C32. https://doi.org/10.1152/ajpcell.00341.2018 |
||||
| 36 Stutts MJ, Schwab JH, Chen MG, Knowles MR, Boucher RC: Effects of Pseudomonas aeruginosa on bronchial epithelial ion transport. Am Rev Respir Dis 1986;134:17-21. | ||||
| 37 Buyck JM, Verriere V, Benmahdi R, Higgins G, Guery B, Matran R, Harvey BJ, Faure K, Urbach V: P. aeruginosa LPS stimulates calcium signaling and chloride secretion via CFTR in human bronchial epithelial cells. J Cyst Fibros 2013;12:60-67. https://doi.org/10.1016/j.jcf.2012.06.007 |
||||
| 38 Schwarzer C, Wong S, Shi J, Matthes E, Illek B, Ianowski JP, Arant RJ, Isacoff E, Vais H, Foskett JK, Maiellaro I, Hofer AM, Machen TE: Pseudomonas aeruginosa Homoserine lactone activates store-operated cAMP and cystic fibrosis transmembrane regulator-dependent Cl- secretion by human airway epithelia. J Biol Chem 2010;285:34850-34863. https://doi.org/10.1074/jbc.M110.167668 |
||||
| 39 Ivonnet P, Salathe M, Conner GE: Hydrogen peroxide stimulation of CFTR reveals an Epac-mediated, soluble AC-dependent cAMP amplification pathway common to GPCR signalling. Br J Pharmacol 2015;172:173-184. https://doi.org/10.1111/bph.12934 |
||||
| 40 Cantin AM, White TB, Cross CE, Forman HJ, Sokol RJ, Borowitz D: Antioxidants in cystic fibrosis. Conclusions from the CF antioxidant workshop, Bethesda, Maryland, November 11-12, 2003. Free Radic Biol Med 2007;42:15-31. https://doi.org/10.1016/j.freeradbiomed.2006.09.022 |
||||
| 41 Lyseng-Williamson KA: Idebenone: A Review in Leber's Hereditary Optic Neuropathy. Drugs 2016;76:805-813. https://doi.org/10.1007/s40265-016-0574-3 |
||||
| 42 Devisscher L, Van Coillie S, Hofmans S, Van Rompaey D, Goossens K, Meul E, Maes L, De Winter H, Van Der Veken P, Vandenabeele P, Berghe TV, Augustyns K: Discovery of Novel, Drug-Like Ferroptosis Inhibitors with in vivo Efficacy. J Med Chem 2018;61:10126-10140. https://doi.org/10.1021/acs.jmedchem.8b01299 |
||||