×
![]()
Corresponding Author: Barbro N. Melgert
Department of Molecular Pharmacology, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, Groningen, 9713AV (The Netherlands)
Tel. +31 50 36 32947, E-Mail b.n.melgert@rug.nl
Osteoprotegerin Expression in Liver is Induced by IL13 through TGFβ
Adhyatmika Adhyatmikaa,b Kurnia S. S. Putric,d Emilia Gorec Keri A. Mangnusa Catharina Reker-Smita Detlef Schuppane,f Leonie Beljaarsc Peter Olingac Barbro N. Melgertg,h
aDepartment of Pharmacokinetics, Toxicology, and Targeting, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, The Netherlands, bDepartment of Pharmaceutics, Faculty of Pharmacy, Universitas Gadjah Mada, Yogyakarta, Indonesia, cDepartment of Pharmaceutical Technology and Biopharmacy, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, The Netherlands, dFaculty of Pharmacy, Universitas Indonesia, Depok, West Java, Indonesia, eInstitute of Translational Immunology, University Medical Center of the Johannes Gutenberg University Mainz, Mainz, Germany, fDivision of Gastroenterology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA, gDepartment of Molecular Pharmacology, Groningen Research Institute of Pharmacy, University of Groningen, Groningen, The Netherlands, hGroningen Research Institute for Asthma and COPD, University Medical Center Groningen, Groningen, The Netherlands
Introduction
Liver fibrosis is a chronic disease induced by long term injury and/or inflammation initiated by virus infections or chemical-induced injury, for example drugs or alcohol [1]. The main pathological characteristic of liver fibrosis is persistent extracellular matrix formation by hepatic stellate cells, which in turn prevents the regrowth of functional hepatocytes [2]. The disease has a high burden as there is no possible therapy to reverse the process when it has fully developed and therefore transplantation is the only option [3].
Transforming growth factor β (TGFβ) has been widely studied for many years as one of the central players in liver fibrosis, but this has not yielded any effective new drugs yet [4, 5]. It is therefore likely that the process of fibrosis development is far more complicated than just the actions of TGFβ alone and that we need to understand the different players and interactions better to develop potential drug candidates.
We recently became interested in the actions of osteoprotegerin (OPG, gene name TNFRSF11B) after finding that OPG is produced in high quantities by (liver) fibroblasts, especially after stimulation with TGFβ and that OPG itself can induce expression of TGFβ, indicating a feed-forward loop [6]. Several clinical studies have shown that higher serum levels of OPG are associated with having liver fibrosis/cirrhosis [7-13]. In addition, OPG serum levels are part of a novel diagnostic score called Coopscore® that has better diagnostic performance than Fibrometer®, Fibrotest®, Hepascore® and Fibroscan™ in chronic hepatitis C-associated fibrosis [8]. Moreover, in our previous studies, we have demonstrated high hepatic OPG production in liver tissue of patients transplanted for liver cirrhosis and in murine models of liver fibrosis.
Osteoprotegerin is well known for its role in protecting bone matrix degradation [14], but little is known about its function in nonbone tissues. In that respect, its role in vascular calcifications is probably best studied, showing that OPG protects against vascular calcification [15]. This contrasts with its known functional influence in bone metabolism in which it induces calcification of bone [14]. This suggests that OPG has more possible functions unrelated to bone and our previous data show its firm associations with fibrotic processes and TGFβ signaling in (myofibroblasts) [6]. However, little is known about the regulation of OPG production in (liver) fibroblasts by other mediators involved in fibrosis [16]. In this study we therefore aimed to further investigate OPG regulation in the liver by studying the effects of several key fibrosis-related growth factors/interleukins and their downstream signaling pathways. These were interleukin (IL) 1β representing a pro-inflammatory and profibrotic mediator, platelet-derived growth factor BB (PDGF-BB), and IL13, both well-known pro-fibrotic mediators for early and late fibrosis respectively.
Materials and Methods
Animals
Male and female wild-type C57BL/6 mice were obtained from Harlan (Horst, The Netherlands) and male STAT6(-/-) C57BL/6 mice were bred in the Institute of Translational Immunology, University Medical Center of the Johannes Gutenberg University Mainz, Germany [17]. Animals were kept in cages with a 12 hour of light/dark cycle and received food and water ad libitum. The use of C57BL/6 mice in this study was approved by the Institutional Animal Care and Use Committee of the University of Groningen (DEC 6416 AA) and the use of STAT6(-/-) mice by the Institutional Animal Care and Use Committee of the Government of Rhineland Palatinate under the reference number 2317707/G12-1-007.
Precision-cut liver slices
Murine precision-cut liver slices were prepared as described before by De Graaf et al. [18]. Slices were treated with 5 ng/mL TGFβ (Peprotech, Rocky Hill, US), 10 ng/mL IL13 (Peprotech), 10 ng/mL IL1β (Peprotech), 10 ng/mL PDGF-BB (Peprotech), 10 mM galunisertib (Selleckchem, Munich, Germany), 21 nM AS1517499 (Axon MedChem, Groningen, The Netherlands), and/or 10 μM T5224 (ApexBio, Houston, US) in triplicate for a total of 48 hours and culture medium was refreshed every 24 hours.
In vitro cell lines
50,000/well 3T3 murine fibroblasts (The American Type Culture Collection, ATCC® CRL-1658) were cultured in standard medium of Gibco® Dulbecco’s Modified Eagle Medium (Thermo Scientific, Waltham, Massachussets, US) containing 4.5 g/L D-Glucose (Sigma-Aldrich, Missouri, US), 2 mM L-Glutamine (Thermo Scientific, Waltham, Massachussets, US), and 10% of fetal calf serum (Biowest, Nuaillé, France). Cells were starved with medium containing 0.5% serum 24 hours prior to other treatments. Treatments with TGFβ, IL13, IL1β, and PDGF-BB were done at similar concentrations as described for the experiments with slices.
Generation of tissue lysate
Tissue slices were lysed with extraction buffer containing 25 mM Tris (Sigma-Aldrich, Missouri, US), 10 mM sodium phosphate (Sigma-Aldrich), 150 mM NaCl (Sigma-Aldrich, Missouri, US), 0.1% SDS (Sigma-Aldrich, Missouri, US), 1% Triton-X 100 (Sigma-Aldrich, Missouri, US), and protease inhibitor (Thermo Scientific, Waltham, Massachussets, US) and incubated for 5 minutes at room temperature before snap-freezing and stored at -80°C until analysis.
Osteoprotegerin analysis
Osteoprotegerin was measured in culture supernatants of cells and slices using a murine OPG DuoSet® ELISA kit (R&D Systems, Minneapolis, US) according to the instructions provided by the manufacturer.
Messenger RNA analysis
Messenger RNA was isolated from cells or slices (three slices per sample, pooled, homogenized prior to extraction) using Maxwell® LEV Simply RNA Cells/Tissue kit (Promega, Madison, Wisconsin, US). A NanoDrop® ND-1000 Spectrophotometer (Thermo Scientific) was used to measure total mRNA concentration in samples. cDNA synthesis from the mRNA was performed using a Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) kit (Promega, Madison, Wisconsin, USA) in a Mastercycler® Gradient (Eppendorf, Hamburg, Germany) programmed for 10 minutes at 20°C, 30 minutes at 42°C, 12 minutes at 20°C, 5 minutes at 990C, and 5 minutes at 20°C. Transforming growth factor beta 1 (TGFβ1), IL13 receptor α2 (IL13Rα2), pro-collagen 1 subunit α1 (Col1α1), α-smooth muscle actin (αSMA), heat shock protein 47 (HSP47), plasminogen activator inhibitor 1 (PAI1), and fibronectin 1 (Fn1) genes were quantified using quantitative real time PCR (RT qPCR) from the synthesized cDNA, using SensiMixTM SYBR® Green (Bioline, London, UK) in a 7900HT Real-Time PCR sequence detection system (Applied Biosystems, Waltham, Massachussets, US) with primer sequences as presented in Table 1. PCR analysis consisted of 45 cycles of 10 min at 95°C, 15 seconds at 95°C, and 25 seconds at 60°C (repeated for 40 times) followed by a dissociation stage of 95°C for 15 seconds, 60°C for 15 seconds, and 95°C for 15 seconds. Output data were analyzed using SDS 2.4 software (Applied Biosystems) and ΔCt values were calculated after β-actin normalization. Two to the power of -ΔCt (2-ΔCt) was used as a final value to be statistically analyzed.
Viability assay
Viability of the slices was assessed by measuring the ATP content per milligram tissue using a bioluminescence assay kit (Sigma-Aldrich) as previously reported by Hadi et al. [19]. For each sample, three slices were collected separately in 1 mL sonification optimization (SONOP) solution pH 10.9 containing 70% ethanol and 2 nM EDTA.
Statistics
All statistics were performed using GraphPad Prism 8. As datasets were all n<8, nonparametric tests were used. When comparing 2 groups a Mann Whitney U or Wilcoxon test was used depending on the data being paired or not. When comparing multiple groups, a Friedman or Kruskall-Wallis with Dunn’s correction was used. Data are presented as min-to-max box-and-whisker plots with individual data points. For the time course experiment using 3T3 fibroblasts, the areas under the curve from 0.5-12 hours and 12-36 hours were calculated and these were compared between groups. Data in this experiment are presented a median + the interquartile range. For all experiments, p<0.05 was considered significant.
Results
IL13 induces fibroblast and hepatic OPG production
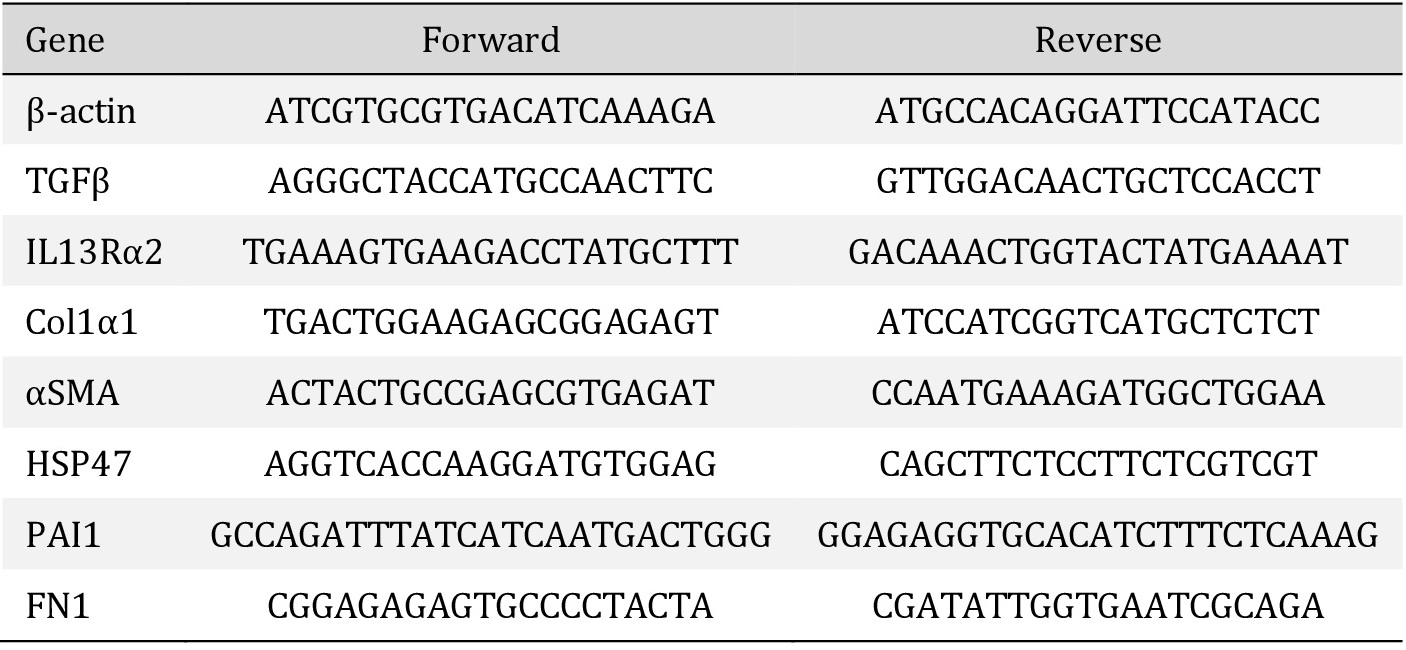
To study possible factors that can induce OPG production by fibroblasts, we treated 3T3 fibroblasts with several cytokines associated with fibrosis. In this study we used a major pro-inflammatory and profibrotic cytokine IL1β, and pro-fibrotic cytokines IL13 and PDGF-BB with TGFβ as our positive control as we have shown higher OPG expression with TGFβ in our previous study [6]. In addition to TGFβ, only IL13 resulted in higher OPG production as compared to control (Fig. 1A). To confirm that IL13 can have a similar effect in liver tissue, we treated murine precision-cut liver slices with IL13 using TGFβ again as a positive control. We have previously shown in multiple publications that TGFβ induces robust fibrotic responses in liver slices of fibrosis-associated markers col1α1, HSP47, FN1, αSMA, and PAI1 on a protein level which tracked well with similar changes on the RNA level [20, 21]. Similar to treatment with TGFβ, treatment IL13 also resulted in significantly higher OPG release from liver tissue as compared to control (Fig. 1B). This higher OPG release in slices was accompanied by near-significant higher OPG mRNA expression and significant higher expression of fibrosis-associated genes col1α1, HSP47, and FN1, but not αSMA and PAI1 (Fig. 1C), suggesting IL13 is less fibrogenic than TGFβ. None of the treatments affected the viability of the slices (Supplementary Fig. S1 – for all supplementary material see www.cellphysiolbiochem.com).
IL13 induces OPG production at a slower rate than TGFβ
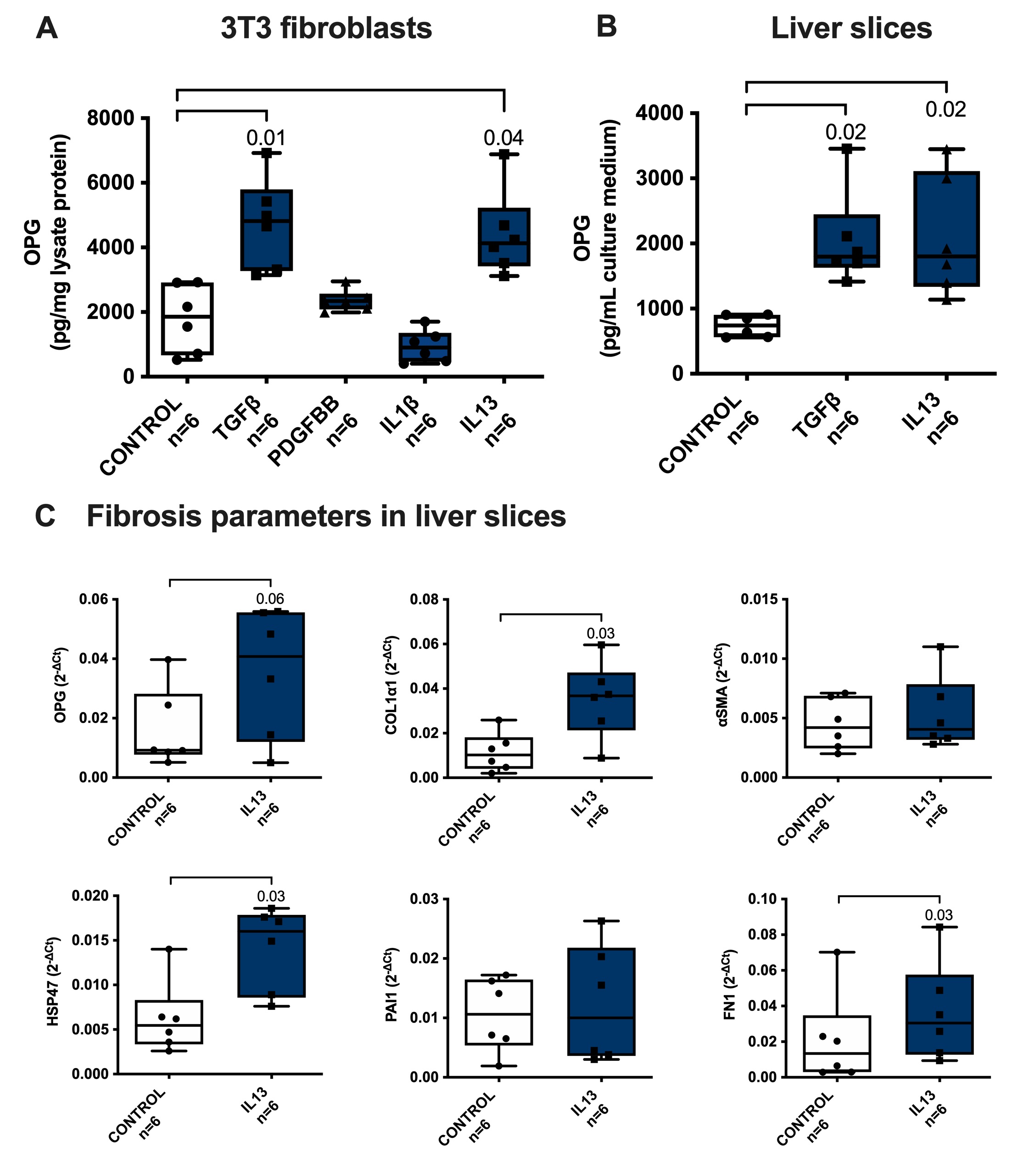
To check whether induction of OPG production followed similar kinetics between TGFβ and IL13, we followed OPG release in time in culture medium of 3T3 fibroblasts after stimulation with TGFβ and IL13. We found that after 36 hours of incubation IL13 and TGFβ both induced a similar release in OPG although the induction by TGFβ occurred somewhat faster. When comparing the area under the curve between stimulated cells and untreated control cells in the first 12 hours, we found a significant increase in OPG release by TGFβ, while IL13 was not significantly different from control. In the time interval from 12 to 36 hours both TGFβ and IL13 significantly induced OPG release as compared to control (Fig. 2).
IL13 induces hepatic OPG induction through TGFβ
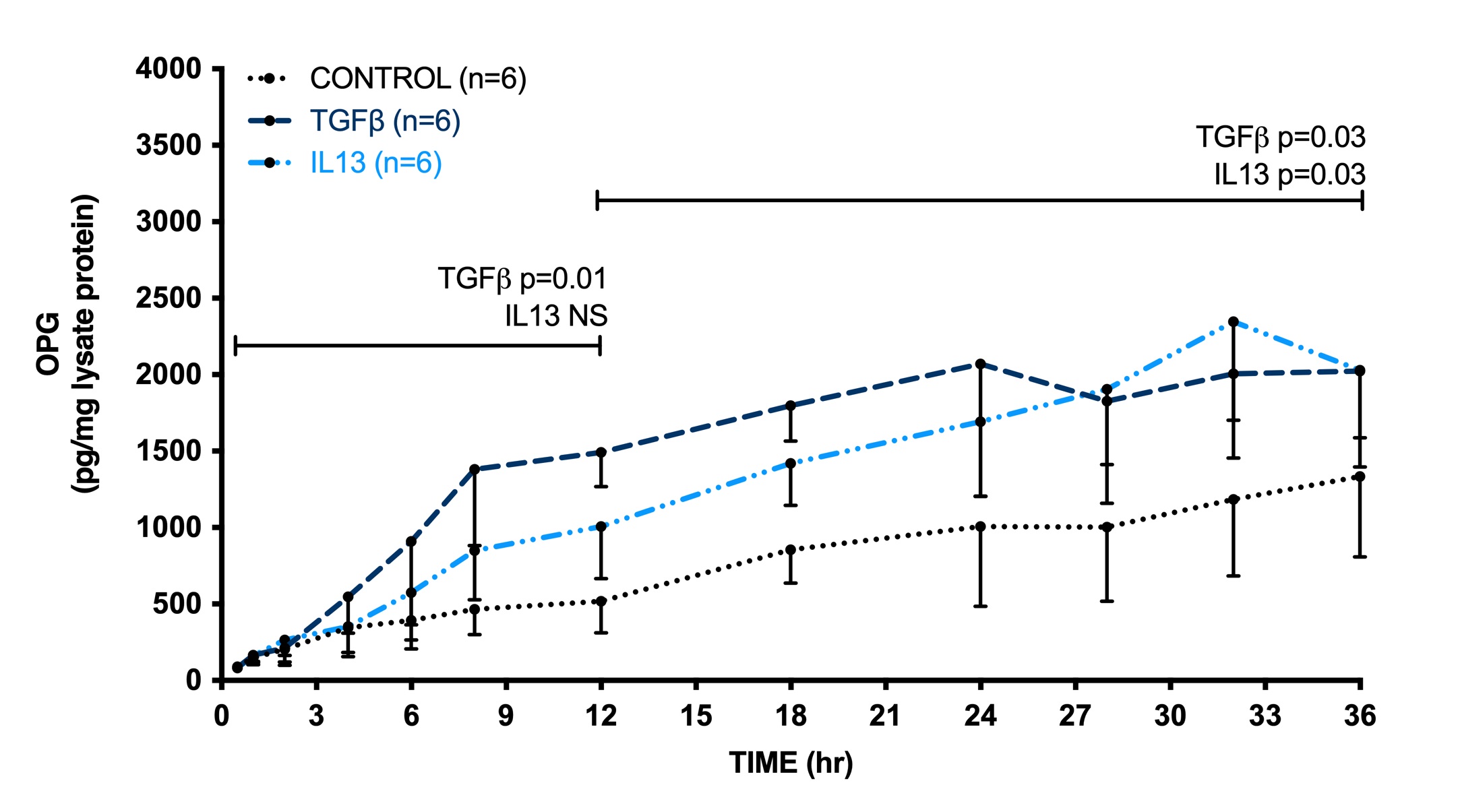
We hypothesized that TGFβ may be involved in the higher hepatic OPG production by mouse liver tissue after IL13 treatment as IL13 has been shown to induce TGFβ1 expression [22]. We therefore assessed TGFβ1 mRNA expression in liver slices after incubation with IL13 and we found a trend towards higher TGFβ1 mRNA expression after IL13 treatment compared to untreated control slices (Fig. 3a). To confirm that TGFβ is indeed involved in the IL13 effect on OPG induction, we also incubated liver slices with galunisertib, a TGFβ1 receptor inhibitor, together with IL13. We found that with galunisertib cotreatment, IL13 treatment did not result in higher OPG release from liver tissue anymore (Fig. 3b). None of the treatments affected the viability of the slices (Supplementary Fig. S1).
STAT6 is involved in IL13-induced release of OPG
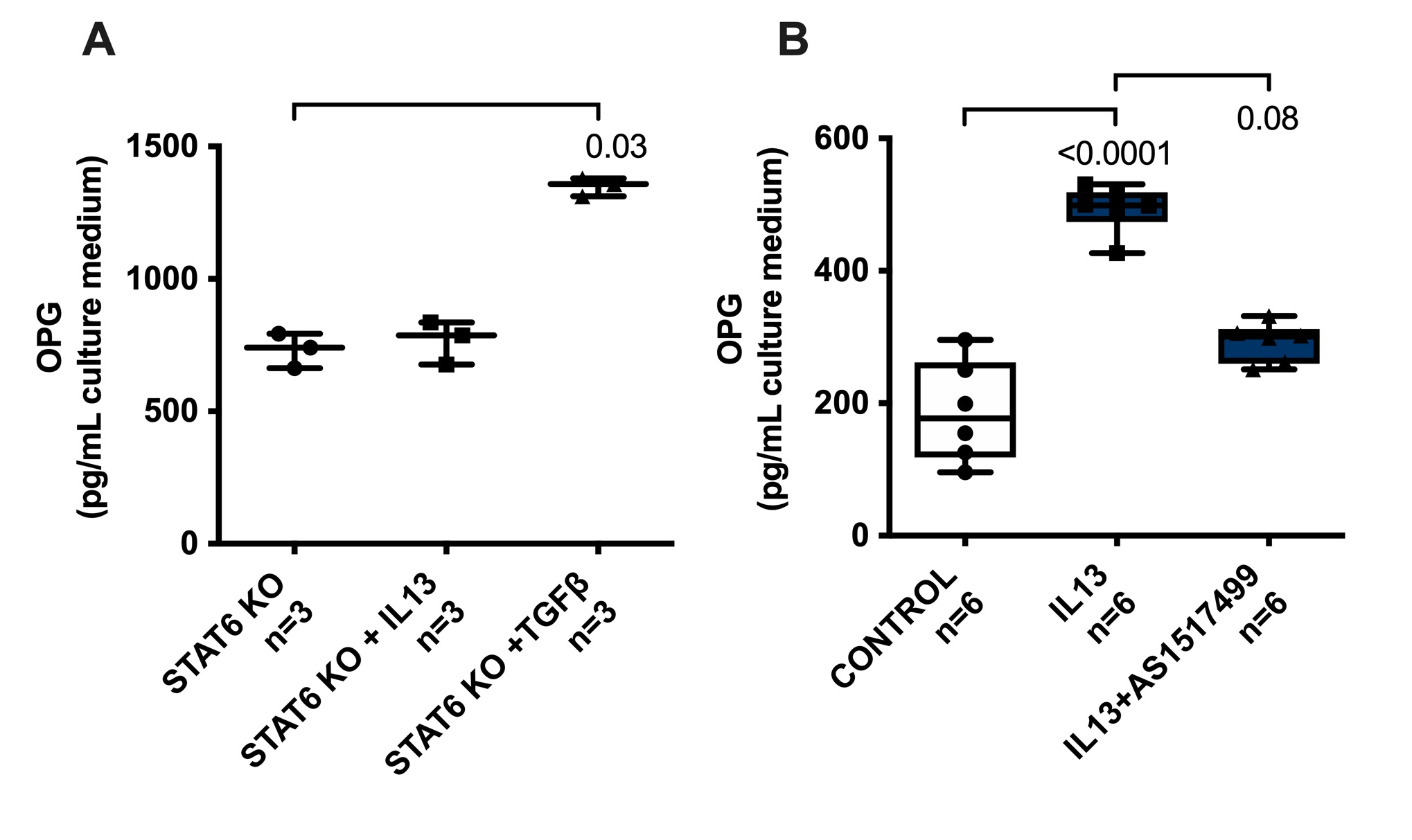
IL13 has been reported to signal through 2 receptors: receptor IL13Rα1 and IL13Rα2 [23]. The downstream activation pathway of IL13Rα1 is via transcription factor STAT6 [24]. To study whether the activation of IL13Rα1 and subsequently STAT6 is involved in the IL13-induced release of OPG, we treated liver slices of STAT6-deficient mouse with IL13 or TGFβ and measured OPG released in medium. We found that IL13 failed to induce OPG release by liver slices of STAT6-deficient mice as compared to untreated controls, whereas TGFβ could still induce OPG release as we found before in wildtype mice (Fig. 4a). To confirm our finding, we used AS1517499, a chemical compound blocking STAT6 activity, in our wild-type mouse liver slices [25] and similarly found that IL13 did not induce OPG release anymore when slices were co-incubated with this inhibitor as compared to slices only treated with IL13 (Fig. 4b). None of the treatments affected the viability of the slices (Supplementary Fig. S1).
IL13 receptor α2 is also involved in IL13-induced OPG release
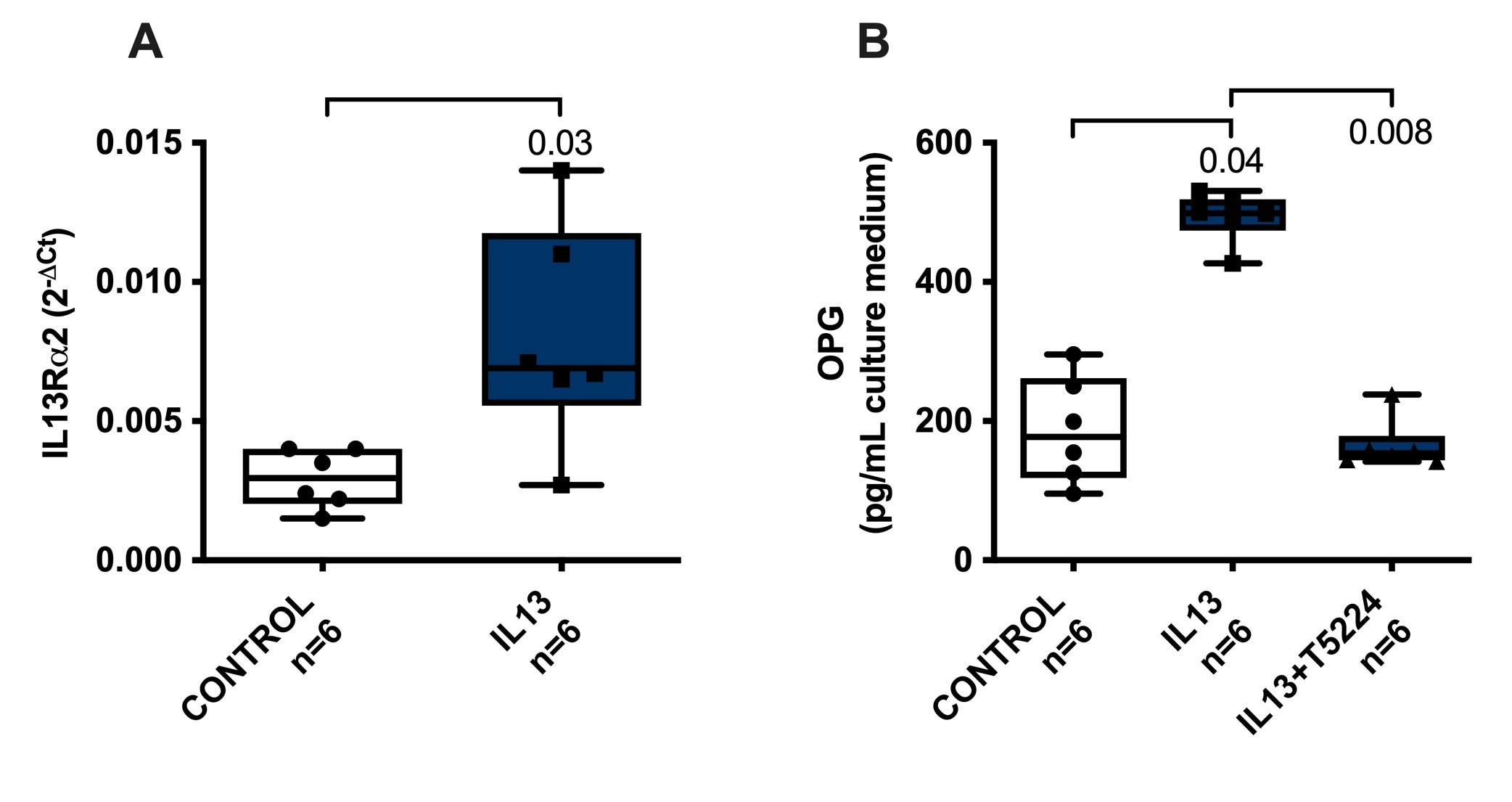
Fichtner-Feigl et al. reported that IL13Rα2 is involved in induction of TGFβ expression and fibrosis through transcription factor AP1 [26]. However, in homeostatic conditions, the expression of this receptor is low [27], while activation of IL13Rα1 and subsequently STAT6 can induce IL13Rα2 expression [28]. In order to check whether these findings are also relevant in our system, we assessed IL13Rα2 mRNA expression in liver slices upon IL13 treatment. We found that IL13Rα2 mRNA expression level was significantly higher upon IL13 treatment as compared to untreated controls (Fig. 5a). We then used T5224, a chemical inhibitor of AP1 [29] to study whether AP1 in involved in IL13-induced OPG release and we found that indeed chemical inhibition of AP1 completely abolished the IL13-induced release of OPG (Fig. 5b). None of the treatments affected the viability of the slices (Supplementary Fig. S1).
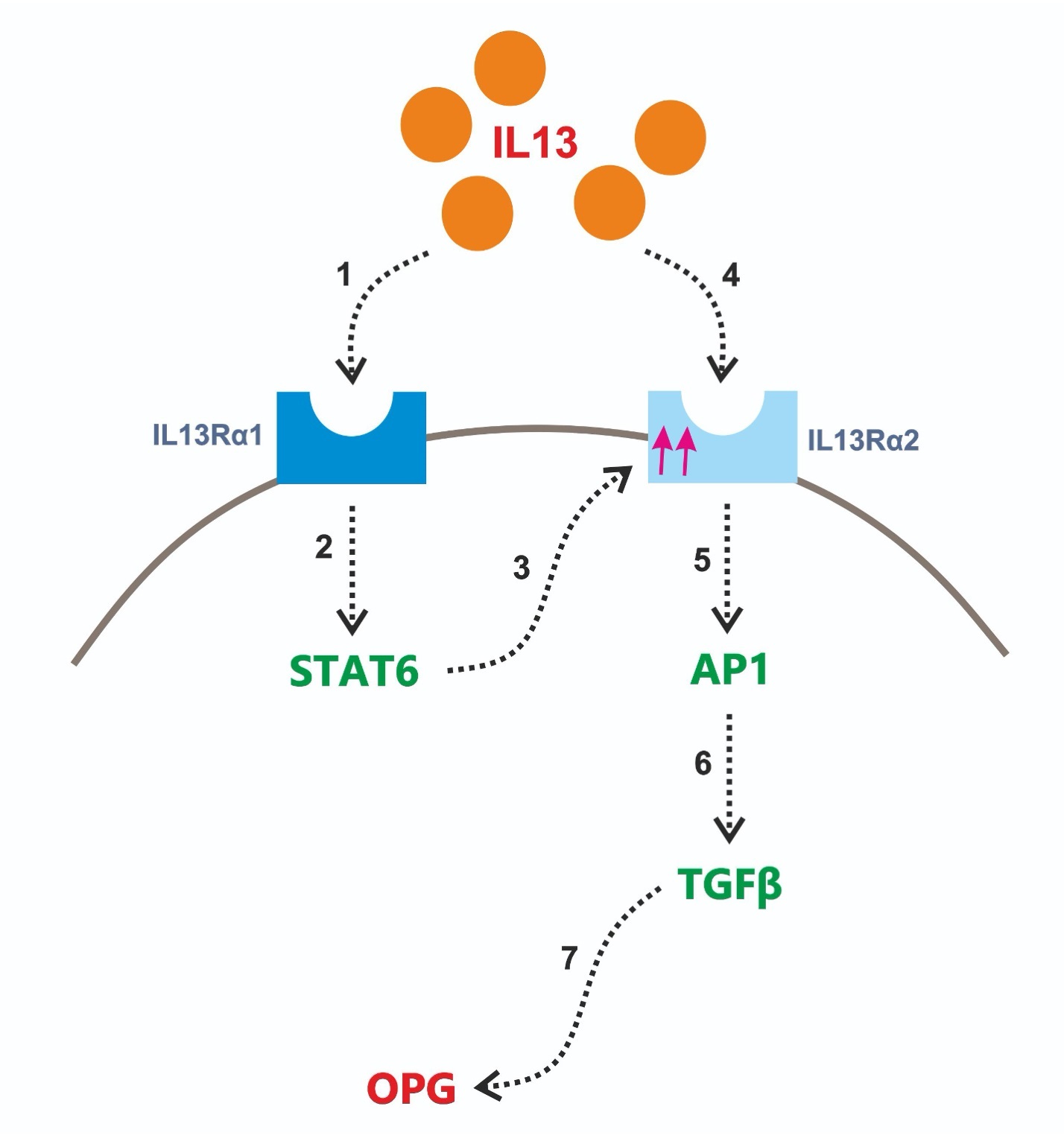
We have shown that IL13 induces OPG release by liver tissue through a TGFβ-dependent pathway involving both the α1 and the α2 receptor of IL13 and transcription factors STAT6 and AP1. OPG may therefore be a novel target for the treatment liver fibrosis as it is mechanistically linked to two important regulators of fibrosis in liver, namely IL13 and TGFβ1.
Author Contributions
Conceptualization, A.A., L.B., and B.N.M., data curation, A.A., K.S.S.P., E.G., K.A.M., C.R.-S., and B.N.M., formal analysis, A.A., L.B., and B.N.M., funding acquisition, P.O., and B.N.M., investigation, A.A., L.B., and B.N.M., methodology, A.A., K.S.S.P., L.B., C.R.-S., D.S., P.O., and B.N.M., project administration, B.N.M., resources, B.N.M., software, B.N.M., supervision, L.B., P.O., and B.N.M., validation, L.B. and B.N.M., visualization, A.A., L.B., and B.N.M., writing—original draft, A.A., K.S.S.P. and B.N.M., writing—review and editing, K.A.M., C.R.-S., D.S., L.B., P.O., and B.N.M. All authors have read and agreed to the published version of the manuscript.
Funding Sources
A.A. received scholarship from LPDP (The Indonesian Endowment Funds for Education, Ministry of Finance, Republic of Indonesia) and K.S.S.P. from DIKTI (The Ministry of Higher Education, Republic of Indonesia) to undergo their Ph.D. education in the Groningen Research Institute of Pharmacy, University of Groningen, The Netherlands. DS received project-related support from EU Horizon 2020 projects under grant agreements nr. 634413 (EPoS, European Project on Steatohepatitis).
Statement of Ethics
The use of C57BL/6 mice in this study was approved by the Institutional Animal Care and Use Committee of the University of Groningen (DEC 6416 AA) and the use of STAT6(-/-) mice by the Institutional Animal Care and Use Committee of the Government of Rhineland Palatinate under the reference number 2317707/G12-1-007.
The authors have no conflicts of interest to declare.
| 1 Friedman SL: Liver fibrosis -- from bench to bedside. J Hepatol 2003;38:S38-S53. https://doi.org/10.1016/S0168-8278(02)00429-4 |
||||
| 2 Bataller R, Brenner DA: Liver fibrosis. J Clin Invest 2005;115:209-218. https://doi.org/10.1172/JCI24282 |
||||
| 3 Ismail MH, Pinzani M: Reversal of liver fibrosis. Saudi J Gastroenterol 2009;15:72-79. https://doi.org/10.4103/1319-3767.45072 |
||||
| 4 Xu F, Liu C, Zhou D, Zhang L: TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem 2016;64:157-167. https://doi.org/10.1369/0022155415627681 |
||||
| 5 Fabregat I, Moreno-Caceres J, Sanchez A, Dooley S, Dewidar B, Giannelli G, Ten Dijke P, Consortium I-L: TGF-beta signalling and liver disease. FEBS J 2016;283:2219-2232. https://doi.org/10.1111/febs.13665 |
||||
| 6 Adhyatmika A, Beljaars L, Putri KSS, Habibie H, Boorsma CE, Reker-Smit C, Luangmonkong T, Guney B, Haak A, Mangnus KA, Post E, Poelstra K, Ravnskjaer K, Olinga P, Melgert BN: Osteoprotegerin is more than a possible serum marker in liver fibrosis: A study into its function in human and murine liver. Pharmaceutics 2020;12:1-21. https://doi.org/10.3390/pharmaceutics12050471 |
||||
| 7 Garcia-Valdecasas-Campelo E, Gonzalez-Reimers E, Santolaria-Fernandez F, De la Vega-Prieto MJ, Milena-Abril A, Sanchez-Perez MJ, Martinez-Riera A, Gomez-Rodriguez Mde L: Serum osteoprotegerin and RANKL levels in chronic alcoholic liver disease. Alcohol Alcohol 2006;41:261-266. https://doi.org/10.1093/alcalc/agl004 |
||||
| 8 Bosselut N, Taibi L, Guechot J, Zarski JP, Sturm N, Gelineau MC, Poggi B, Thoret S, Lasnier E, Baudin B, Housset C, Vaubourdolle M, Group AHF: Including osteoprotegerin and collagen IV in a score-based blood test for liver fibrosis increases diagnostic accuracy. Clin Chim Acta 2013;415:63-68. https://doi.org/10.1016/j.cca.2012.09.020 |
||||
| 9 Prystupa A, Dabrowska A, Sak JJ, Tarach J, Torun-Jurkowska A, Lachowska-Kotowska P, Dzida G: Concentrations of fetuin-A, osteoprotegerin and alpha-Klotho in patients with alcoholic liver cirrhosis. Exp Ther Med 2016;12:3464-3470. https://doi.org/10.3892/etm.2016.3754 |
||||
| 10 Monegal A, Navasa M, Peris P, Alvarez L, Pons F, Rodes J, Guanabens N: Serum osteoprotegerin and its ligand in cirrhotic patients referred for orthotopic liver transplantation: relationship with metabolic bone disease. Liver Int 2007;27:492-497. https://doi.org/10.1111/j.1478-3231.2007.01448.x |
||||
| 11 Fabrega E, Orive A, Garcia-Suarez C, Garcia-Unzueta M, Antonio Amado J, Pons-Romero F: Osteoprotegerin and RANKL in alcoholic liver cirrhosis. Liver Int 2005;25:305-310. https://doi.org/10.1111/j.1478-3231.2005.01073.x |
||||
| 12 Szalay F, Hegedus D, Lakatos PL, Tornai I, Bajnok E, Dunkel K, Lakatos P: High serum osteoprotegerin and low RANKL in primary biliary cirrhosis. J Hepatol 2003;38:395-400. https://doi.org/10.1016/S0168-8278(02)00435-X |
||||
| 13 Moschen AR, Kaser A, Stadlmann S, Millonig G, Kaser S, Muhllechner P, Habior A, Graziadei I, Vogel W, Tilg H: The RANKL/OPG system and bone mineral density in patients with chronic liver disease. J Hepatol 2005;43:973-983. https://doi.org/10.1016/j.jhep.2005.05.034 |
||||
| 14 Boyce BF, Xing L: Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther 2007;9:S1. https://doi.org/10.1186/ar2165 |
||||
| 15 Harper E, Forde H, Davenport C, Rochfort KD, Smith D, Cummins PM: Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vascul Pharmacol 2016;82:30-40. https://doi.org/10.1016/j.vph.2016.02.003 |
||||
| 16 Wallace K, Burt AD, Wright MC: Liver fibrosis. Biochem J 2008;411:1-18. https://doi.org/10.1042/BJ20071570 |
||||
| 17 Akimoto T, Numata F, Tamura M, Takata Y, Higashida N, Takashi T, Takeda K, Akira S: Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. J Exp Med 1998;187:1537-1542. https://doi.org/10.1084/jem.187.9.1537 |
||||
| 18 de Graaf IA, Olinga P, de Jager MH, Merema MT, de Kanter R, van de Kerkhof EG, Groothuis GM: Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc 2010;5:1540-1551. https://doi.org/10.1038/nprot.2010.111 |
||||
| 19 Hadi M, Chen Y, Starokozhko V, Merema MT, Groothuis GM: Mouse precision-cut liver slices as an ex vivo model to study idiosyncratic drug-induced liver injury. Chem Res Toxicol 2012;25:1938-1947. https://doi.org/10.1021/tx300248j |
||||
| 20 Bigaeva E, Gore E, Simon E, Zwick M, Oldenburger A, de Jong KP, Hofker HS, Schleputz M, Nicklin P, Boersema M, Rippmann JF, Olinga P: Transcriptomic characterization of culture-associated changes in murine and human precision-cut tissue slices. Arch Toxicol 2019;93:3549-3583. https://doi.org/10.1007/s00204-019-02611-6 |
||||
| 21 Bigaeva E, Gore E, Mutsaers HAM, Oosterhuis D, Kim YO, Schuppan D, Bank RA, Boersema M, Olinga P: Exploring organ-specific features of fibrogenesis using murine precision-cut tissue slices. Biochim Biophys Acta Mol Basis Dis 2020;1866:165582. https://doi.org/10.1016/j.bbadis.2019.165582 |
||||
| 22 Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, Senior RM, Elias JA: Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 2001;194:809-821. https://doi.org/10.1084/jem.194.6.809 |
||||
| 23 Chomarat P, Banchereau J: Interleukin-4 and interleukin-13: their similarities and discrepancies. Int Rev Immunol 1998;17:1-52. https://doi.org/10.3109/08830189809084486 |
||||
| 24 Murata T, Husain SR, Mohri H, Puri RK: Two different IL-13 receptor chains are expressed in normal human skin fibroblasts, and IL-4 and IL-13 mediate signal transduction through a common pathway. Int Immunol 1998;10:1103-1110. https://doi.org/10.1093/intimm/10.8.1103 |
||||
| 25 Chiba Y, Todoroki M, Nishida Y, Tanabe M, Misawa M: A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am J Respir Cell Mol Biol 2009;41:516-524. https://doi.org/10.1165/rcmb.2008-0163OC |
||||
| 26 Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A: IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med 2006;12:99-106. https://doi.org/10.1038/nm1332 |
||||
| 27 Daines MO, Tabata Y, Walker BA, Chen W, Warrier MR, Basu S, Hershey GK: Level of expression of IL-13R alpha 2 impacts receptor distribution and IL-13 signaling. J Immunol 2006;176:7495-7501. https://doi.org/10.4049/jimmunol.176.12.7495 |
||||
| 28 David M, Ford D, Bertoglio J, Maizel AL, Pierre J: Induction of the IL-13 receptor alpha2-chain by IL-4 and IL-13 in human keratinocytes: involvement of STAT6, ERK and p38 MAPK pathways. Oncogene 2001;20:6660-6668. https://doi.org/10.1038/sj.onc.1204629 |
||||
| 29 Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, Hirono S, Shiozawa S: Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol 2008;26:817-823. https://doi.org/10.1038/nbt1412 |
||||
| 30 Firszt R, Francisco D, Church TD, Thomas JM, Ingram JL, Kraft M: Interleukin-13 induces collagen type-1 expression through matrix metalloproteinase-2 and transforming growth factor-beta1 in airway fibroblasts in asthma. Eur Respir J 2014;43:464-473. https://doi.org/10.1183/09031936.00068712 |
||||
| 31 Weng SY, Wang X, Vijayan S, Tang Y, Kim YO, Padberg K, Regen T, Molokanova O, Chen T, Bopp T, Schild H, Brombacher F, Crosby JR, McCaleb ML, Waisman A, Bockamp E, Schuppan D: IL-4 Receptor Alpha Signaling through Macrophages Differentially Regulates Liver Fibrosis Progression and Reversal. EBioMedicine 2018;29:92-103. https://doi.org/10.1016/j.ebiom.2018.01.028 |
||||
| 32 Gieseck RL, 3rd, Ramalingam TR, Hart KM, Vannella KM, Cantu DA, Lu WY, Ferreira-Gonzalez S, Forbes SJ, Vallier L, Wynn TA: Interleukin-13 Activates Distinct Cellular Pathways Leading to Ductular Reaction, Steatosis, and Fibrosis. Immunity 2016;45:145-158. https://doi.org/10.1016/j.immuni.2016.06.009 |
||||
| 33 Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214:199-210. https://doi.org/10.1002/path.2277 |
||||
| 34 Bonner JC: Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 2004;15:255-273. https://doi.org/10.1016/j.cytogfr.2004.03.006 |
||||
| 35 Szabo G, Csak T: Inflammasomes in liver diseases. J Hepatol 2012;57:642-654. https://doi.org/10.1016/j.jhep.2012.03.035 |
||||
| 36 Sugimoto R, Enjoji M, Nakamuta M, Ohta S, Kohjima M, Fukushima M, Kuniyoshi M, Arimura E, Morizono S, Kotoh K, Nawata H: Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells. Liver Int 2005;25:420-428. https://doi.org/10.1111/j.1478-3231.2005.01087.x |
||||
| 37 Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, Puri RK: Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J Immunol 2008;181:4656-4665. https://doi.org/10.4049/jimmunol.181.7.4656 |
||||
| 38 Lin J, Lu F, Zheng W, Xu S, Tai D, Yu H, Huang Z: Assessment of liver steatosis and fibrosis in rats using integrated coherent anti-Stokes Raman scattering and multiphoton imaging technique. J Biomed Opt 2011;16:116024. https://doi.org/10.1117/1.3655353 |
||||
| 39 Korenblat P, Kerwin E, Leshchenko I, Yen K, Holweg CTJ, Anzures-Cabrera J, Martin C, Putnam WS, Governale L, Olsson J, Matthews JG: Efficacy and safety of lebrikizumab in adult patients with mild-to-moderate asthma not receiving inhaled corticosteroids. Respir Med 2018;134:143-149. https://doi.org/10.1016/j.rmed.2017.12.006 |
||||
| 40 Akhurst RJ, Hata A: Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 2012;11:790-811. https://doi.org/10.1038/nrd3810 |
||||
| 41 Schuppan D, Kim YO: Evolving therapies for liver fibrosis. J Clin Invest 2013;123:1887-1901. https://doi.org/10.1172/JCI66028 |
||||
| 42 Guttman-Yassky E, Blauvelt A, Eichenfield LF, Paller AS, Armstrong AW, Drew J, Gopalan R, Simpson EL: Efficacy and Safety of Lebrikizumab, a High-Affinity Interleukin 13 Inhibitor, in Adults With Moderate to Severe Atopic Dermatitis: A Phase 2b Randomized Clinical Trial. JAMA Dermatol 2020;156:411-420. https://doi.org/10.1001/jamadermatol.2020.0079 |
||||
| 43 Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taieb A, Owen R, Putnam W, Castro M, DeBusk K, Lin CY, Voulgari A, Yen K, Omachi TA: Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: A randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol 2018;78:863-871.e11. https://doi.org/10.1016/j.jaad.2018.01.017 |
||||