×
![]()
Corresponding Author: Germán Esteban González
Pontificia Universidad Católica Argentina (BIOMED, UCA-CONICET),
Laboratorio de Patología Cardiovascular Experimental e Hipertensión Arterial,
Alicia Moreau de Justo 1600-Piso 3-(C1107AFF), Buenos Aires (Argentina)
E-Mail germangonzalez@uca.edu.ar
Genetic Deletion of Galectin-3 Exacerbates Age-Related Myocardial Hypertrophy and Fibrosis in Mice
Florencia Sofia Fontana Esteveza
Maria Celeste Betazzaa
Verónica Miksztowicza,b
Ignacio Miguel Seropiana,c
Mauro Gastón Silvad
Federico Penase
Vanessa Toucedaa,b
Carolina Selsera
Alejo Villaverdea
Nora Gorene
Tomás Francisco Cianciullif
Vanina Medinag
Celina Moralesh
Mariela Gironaccid
Germán Esteban Gonzáleza,i
aPontificia Universidad Católica Argentina, Facultad de Medicina, Instituto de Investigaciones Biomédicas UCA-CONICET, Laboratorio de Patología Cardiovascular Experimental e Hipertensión Arterial, Buenos Aires, Argentina, bUniversidad de Buenos Aires, Facultad de Odontología, Cátedra de Bioquímica General y Bucal, Buenos Aires, Argentina, cServicio de Hemodinamia y Cardiología Intervencionista, Hospital Italiano de Buenos Aires, Argentina, dUniversidad de Buenos Aires, Facultad de Farmacia y Bioquímica, Departamento Química Biológica, IQUIFIB (UBA-CONICET), Buenos Aires, Argentina, eInstituto de Investigaciones Biomédicas en Retrovirus y SIDA, CONICET-Universidad de Buenos Aires, Buenos Aires, Argentina, fDivisión Cardiología, Hospital General de Agudos “Dr. Cosme Argerich”, Buenos Aires, Argentina, gPontificia Universidad Católica Argentina, Facultad de Medicina, Instituto de Investigaciones Biomédicas UCA-CONICET, Laboratorio de Biología Tumoral e Inflamación, Buenos Aires, Argentina, hUniversidad de Buenos Aires, Facultad de Medicina, Departamento de Patología, Buenos Aires, Argentina, iUniversidad de Buenos Aires, Facultad de Odontología, Cátedra de Anatomía Patológica, Buenos Aires, Argentina
Introduction
Increases in lifespan and an aging population worldwide have amplified the prevalence of chronic diseases and placed heightened importance on expanding the understanding of the increasingly prevalent cardiovascular diseases [1]. Aging is a complex and time-dependent physiological process associated with progressive decline and loss of a variety of physiological cell functions, leading to an increased risk of health complications [2, 3]. Aging notably affects the cardiovascular system, significantly increasing the risk of heart disease and the development of heart failure. Studying the underlying mechanisms associated with cardiac aging thus has relevant clinical implications [1, 4].
Constitutive aging of the heart increases its susceptibility to stress and contributes to elevated cardiovascular morbidity and mortality in the elderly [1]. The hallmark of cardiac aging is a progressive and adverse remodeling characterized by myocardial hypertrophy, inflammation, fibrosis, apoptosis, and dysfunction due to a wide variety of interconnected factors promoted by aging. Angiotensin II (Ang II), the main effector peptide of the Renin Angiotensin System (RAS), has been highly implicated in cardiac remodeling and it is recognized to play an important contribution to aged-related myocardial hypertrophy and fibrosis [5]. Ang-(1-7), converted from Ang II by angiotensin converting enzyme 2 (ACE-2) and considered an antagonist of Ang II, attenuated the myocardial hypertrophy and fibrosis in Ang II induced hypertension [6] and diminished the sarcopenia and osteoporosis in old mice [7] but its role in cardiac aging is less known. Likewise, chronic pharmacologic inhibition of RAS markedly prevented the adverse cardiac remodeling associated to aging and significantly enhanced the longevity [8]. In addition, other factors such as transforming growth factor-β (TGF-β), metalloproteinase (MMP), and sirtuins (SIRT) [9–13] and closely linked to the RAS are important contributors to morphological and functional changes of cardiac aging.
Over the last few years, clinical and experimental studies have strongly suggested that galectin-3 (Gal-3), a β-galactosidase–binding lectin, is highly increased in a variety of pathological conditions and promotes cardiac remodeling by regulating the underlying mechanisms of myocardial hypertrophy, inflammation, and fibrosis [14–19]. Additionally, Gal-3 has been found to be an independent prognostic factor for heart failure [14, 15, 20, 21]. Although the role of Gal-3 in cardiovascular pathology appears to be clearly defined, the involvement of Gal-3 in age-related cardiac remodeling is still unknown. Gal-3 regulates cell growth and proliferation as well as apoptosis by orchestrating cell–cell and cell–extracellular matrix interactions [14, 22]. Gal-3 was found to be increased in elderly patients and was proposed as a biomarker associated with frailty in subjects with systolic heart failure [23, 24]. In addition, Gal-3 was associated with elevated left ventricular (LV) mass in age- and sex-matched analysis, suggesting a role in age-related cardiac remodeling [25]. A lack of Gal-3 in old mice with exacerbated renal fibrosis, oxidative stress, and renal dysfunction suggests that an absence of Gal-3 during aging amplifies age-related organ damage [26]. However, the role of Gal-3 in age-induced cardiac remodeling is unknown. Here we hypothesized that genetic deletion of Gal-3 exacerbates the age-related myocardial hypertrophy, fibrosis and apoptosis through the mechanisms involving ANGII, TGF-β, MMP-9 and SIRT and, independently of blood pressure.
Materials and Methods
Mice and experimental design
Male C57BL/6J (C57, n=24) and Gal-3 Knockout (Gal-3 KO; n=29) mice were followed up for 24 months from weaning. Animals maintained the light/dark cycle of 12 h with access to water and food ad libitum grouped with 4 animals per cage. Mice were obtained and followed up in our Bioresources facilities at the Biomedical Research Institute (BIOMED UCA-CONICET) of the Pontificia Universidad Católica Argentina. The experimental protocol was approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL) of BIOMED in line with the NIH’s Guide for the Care and Use of Laboratory Animals [27].
Water and food intake and body weight
Body weight, food and water intake were monitored weekly throughout the experimental period. Body weight (gr) evolution per animal was recorded and averaged for each group.
Systolic Blood Pressure
At 24 months of follow-up the animals were trained for one week for the measurement of blood pressure by plethysmography according to the tail cuff method [19]. After training, systolic blood pressure (SBP) values were recorded for three consecutive days. Measurements were made at the same time for each group in a quiet environment and avoiding stressful conditions for the animals. To facilitate statistical analysis and data interpretation, the results of the three measurements were averaged and a single SBP value at 24 months was considered for each animal.
Echocardiography
Age-related cardiac remodeling and function were studied at 24 months by two-dimensional echocardiography. All Two-dimensional echocardiograms were performed using a Vivid 7 machine (General Electric Medical Systems, Horten, Norway) with a phased array 10 MHz transducer. Mice were slightly anesthetized with tribromoethanol (Avertin); 1.15 ml/kg. Briefly, the heart was visualized in the long axis parasternal view for M-mode. Left ventricle (LV) dimension measurement and posterior wall and left ventricular (LV) end-diastolic and end-systolic diameters (LVEDD and LVESD) and LV-posterior wall thicknesses (PWT) and anterior wall thickness (AWT) were measured in systole and diastole. LV mass and fractional shortening (FS) were calculated as described elsewhere [18] as follows:
1) LV mass = 1.055 [(AWT + LVEDD + PWT)3 - (LVEDD)3]
2) FS (%) = [(LVEDD - LVESD)/LVEDD] x 100
Necropsy
At 24 months, the animals were euthanized with an overdose of sodium pentobarbital (150 mg/kg). After euthanasia, hearts, lungs, kidneys and spleens were removed and weighed and tibia lengths were measured. Organ weights were normalized to tibia length or body weight. In order to corroborate the age-cardiac remodeling at 24 months, heart weight, cardiac mass index and blood pressure were matched between young and old animals (Supplementary Fig. 1 – for all supplementary material see www.cellphysiolbiochem.com).
Histology
Hearts were harvested and fixed in formaldehyde. After that, hearts were cut from apex to base, and paraffin embedded. Serial cuts were performed and stained with hematoxilin-eosin (H&E) for measuring myocyte cross-sectional area (MCSA) and Picrosirius red for quantifying myocardial interstitial collagen by using the image-analysis software (ImageJ). In images obtained with light microscope outlines of myocytes were traced, and cell areas were measured by using an image analyzer-software (Image J) [18].
The percentage of collagen for each region was measured with picrosirius red staining, and calculated as: collagen area/tissue area of the high-power field (400 X) as described previously [19].
Cardiac expression of ANG II and ANG-(1-7)
Cardiac Ang II, and Ang-(1-7) expression was measured by radioimmunoassay. Briefly, cardiac samples were homogenized in acid ethanol (0.1 mol/L HCl/80 % ethanol) containing 0.44 mmol/L o-phenanthroline, 1 mmol/L Na+ para-chloromercuribenzoate, 0.12 mmol/L pepstatin A and 25 mmol/L EDTA. Homogenates were centrifuged at 20000 g for 30 min at 4 °C. Proteins in the supernatant were quantified. The supernatant was subsequently lyophilized and Ang extraction and recovery was performed as described elsewhere [28]. Each sample was corrected for each recovery. Ang II level was quantified by radioimmunoassay using Ang-labelled as described previously. Results were expressed as pg/mg tissue [28].
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
mRNA expression was determined using 5× HOT FIREPOL EVAGREEN qPCR (Solis BioDyne, Estonia) in a StepOnePlus Real-Time PCR System. Parameters were as follows: 52 °C for 2 min, 95 °C for 15 min, and 40 cycles at 95 °C for 15 sec, 63 °C (for TGF-β) 62 °C (for b-actin and MMP-9), 57 °C (for SIRT7) or 56 °C (for SIRT1), for 30 sec and 72 °C for 1 min. Normalization was carried out using actin RNA. Quantification was performed using the comparative threshold cycle (Ct) method, as all the primer pairs (target gene/reference gene) were amplified using comparable efficiencies (relative quantity, 2-ΔΔCt) [29, 30]. Primer sequences:
b-Actin: forward: 5′-GGCTGTATTCCCCTCCATCG-3′, reverse: 5′-CCAGTTGGTAACAATGCCATGT-3′
SIRT1: forward: 5′-GCGGCTGACGACTTC-3′, reverse: 5′-GCTGGCGTGTGACGTTC-3′
SIRT7: forward: 5′-GCCGAGAGCGAGCT-3′, reverse: 5′-GCCCGTGTAGACAACCA-3′
MMP-9: forward: 5′-CAGACCAAGGGTACAGCCTGTT-3′, reverse: 5′-AGTGCATGGCCGAACTC-3′
TGF-β: forward: 5′-CACCGGAGAGCCCTGGATA-3′, reverse: 5′-TGTACAGCTGCCGCACACA-3′
Apoptosis determinations
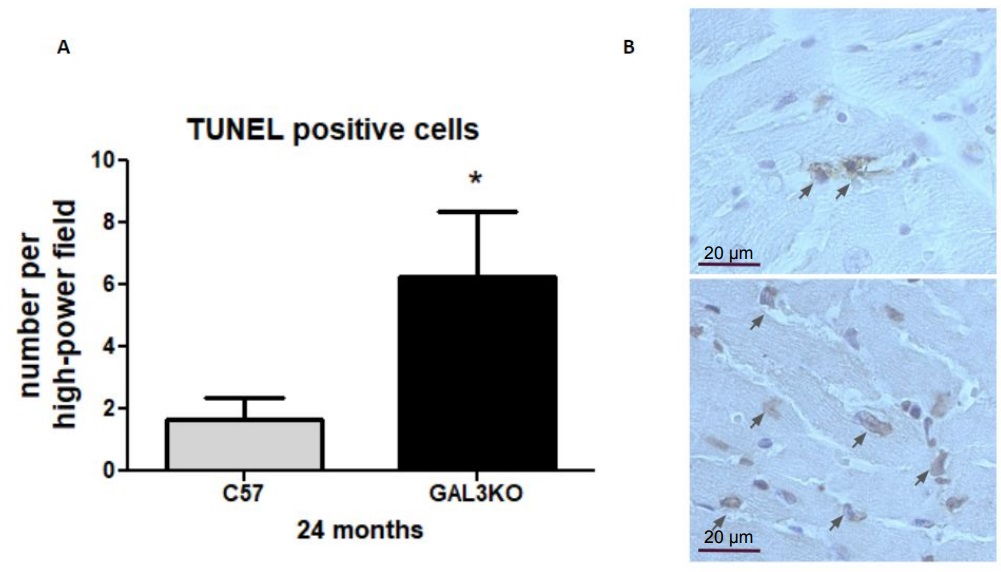
The fragmented DNA in cells undergoing apoptosis was detected in slices from middle sections of the hearts using ApoptagTM plus peroxidase in situ apoptosis Detection Kit (Millipore, MA, USA), according to the manufacturer’s instructions. Tunel-positive cells were quantified in at least 10 high power field (HPF) and the results expressed as Tunel + cells/HPF at 400x magnification.
Statistical analysis
Continuous variables are presented as means ± SEM. Parametric data were analyzed using Student’s unpaired t-test to compare the two groups using Prism 6.0 (GraphPad Software, San Diego, CA). Survival analysis was performed by the Kaplan–Meier method with the log-rank test. Values of p< 0.05 were considered statistically significant.
Results
Decreased survival rate in aged Gal-3 KO mice
During the follow-up survival rate was significantly lower in Gal-3 KO mice compared with C57 mice (p < 0.05 Gal-3 KO vs C57) (Fig. 1A). At 24 months 67 % of C57 (n= 16/24) and 55 % (n= 16/29) of Gal3-KO mice achieved 24 months of age. There were no differences in body weight over the study period (Fig. 1B).
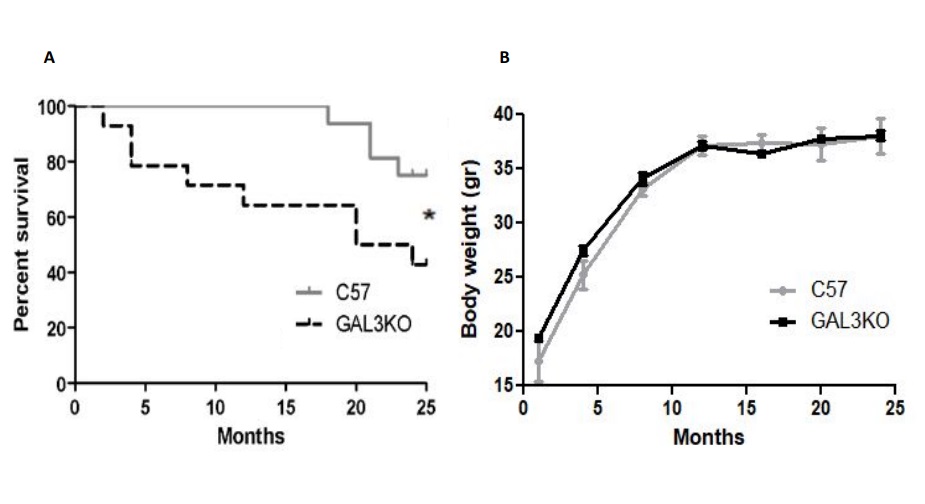
Exacerbated myocardial hypertrophy in aged Gal-3 KO mice independent of blood pressure
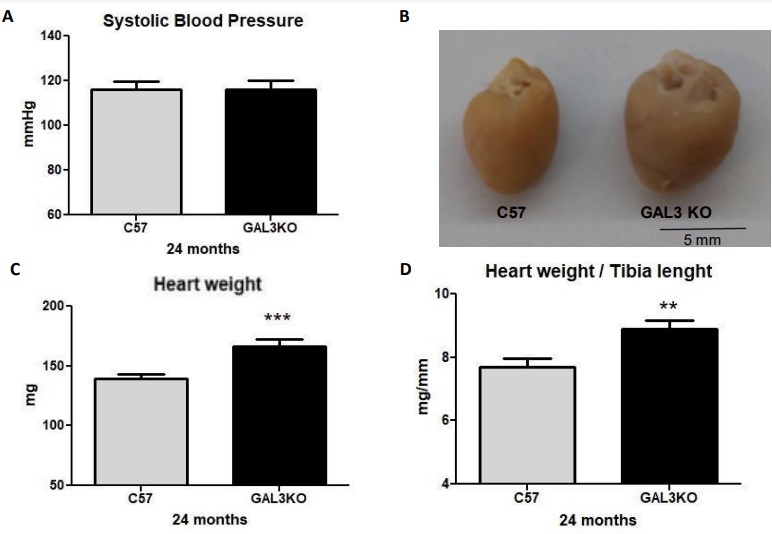
SBP (mmHg) at 24 months was similar between groups and within physiological values (Fig. 2A) while comparing with previous information of young animals, no significant changes were observed between young and aged animals in both groups (Supplementary Fig. 1). Cardiac weight, heart weight/body weight ratio and heart weight/tibia length ratio were significantly increased with aging in both groups (Fig. 2C-D) (Supplementary Fig. 1). However, the increases in cardiac weight and cardiac mass index were higher in aged Gal-3 KO mice than in C57 mice (Fig. 2B-D; p < 0.05). Gal-3 KO mice demonstrated increased renal hypertrophy expressed as kidney weight/tibia length (mg/mm) ratio (27 ± 6 in C57 vs 37 ± 4 in Gal-3 KO; p < 0.05). Additionally, the lung weight/ tibia length (mg/mm) ratio was similar between groups (14.21 ± 5.93 in C57 and 13.69 ± 4.58 in Gal-3 KO mice; p=ns). Spleen weight/ tibia length (mg/mm) were similar in both genotypes (7.13 ± 3.12 in C57 and 6.35 ± 2.21 in Gal-3 KO mice; p=ns).
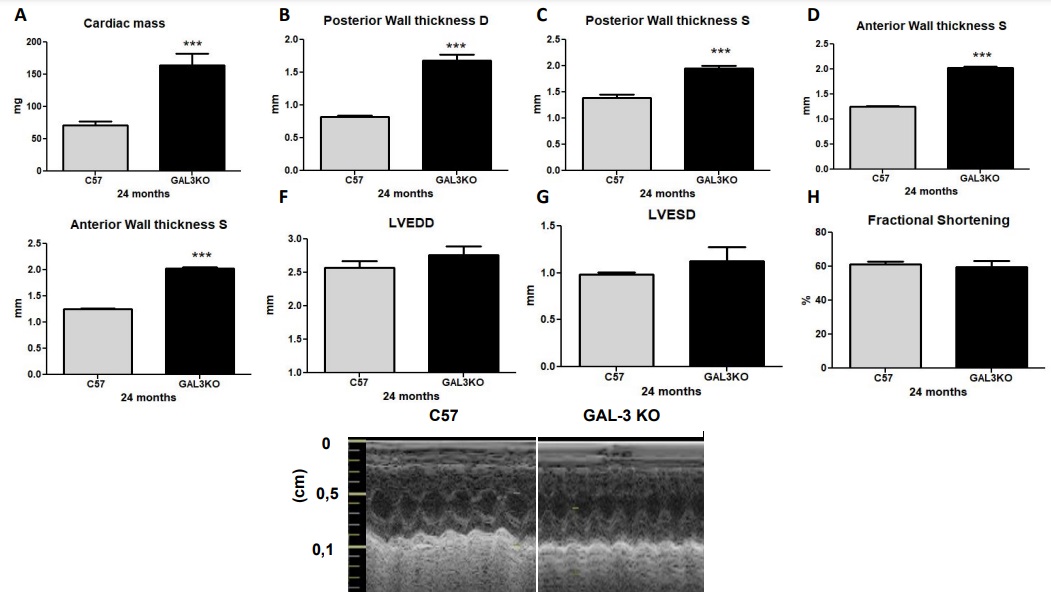
Myocardial hypertrophy in aged Gal-3 KO mice with preserved systolic function
At 24 months, cardiac mass index as calculated by echocardiography was significantly increased in the Gal-3 KO group compared with the C57 group (Fig. 3A; p < 0.01). Additionally, posterior wall thickness and anterior wall thickness in both systole and diastole were significantly increased in the Gal-3 KO group compared to the C57 group (Fig. 3B and C; p < 0.0001). LV end-systolic dimensions (LVESD) and LV end-diastolic dimensions (LVEDD), as well as shortening fraction, were similar between genotypes (Fig. 3D–G; p = NS).
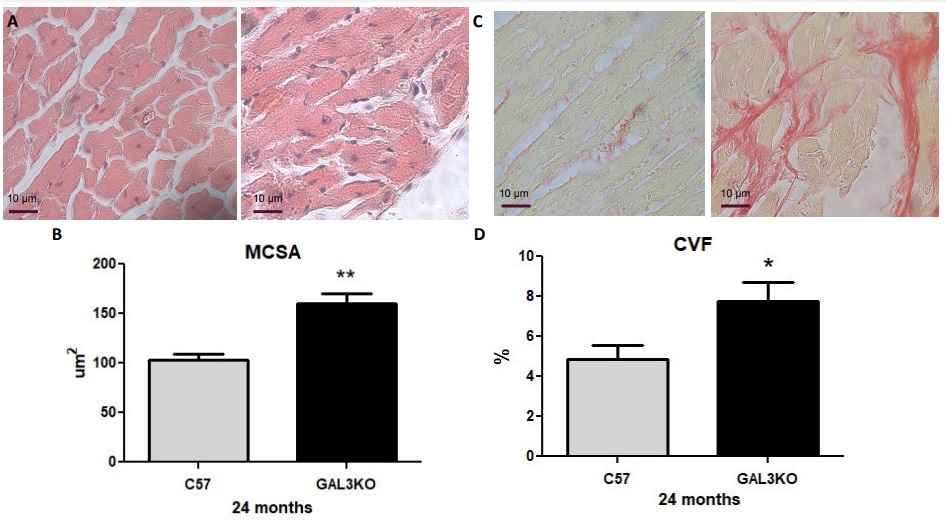
Increased MCSA, fibrosis, and apoptosis in aged Gal-3 KO mice
In order to further confirm that Gal-3 deletion induced cardiac hypertrophy, MCSA was measured at 24 months in Gal-3 KO and control mice. As shown in Fig. 4A, the MCSA in aged Gal-3 KO mice was significantly elevated compared to control mice (p < 0.05). Similarly, interstitial myocardial fibrosis was significantly increased in aged Gal-3 KO mice compared to control mice (Fig. 4B; p < 0.02).
Finally, we quantified myocardial apoptosis as the quantity of TUNEL+ cells and observed that aged Gal-3 KO mice presented with significantly higher numbers of TUNEL+ cells than aged C57 mice (Fig. 5; p < 0.05), indicating increased apoptosis.
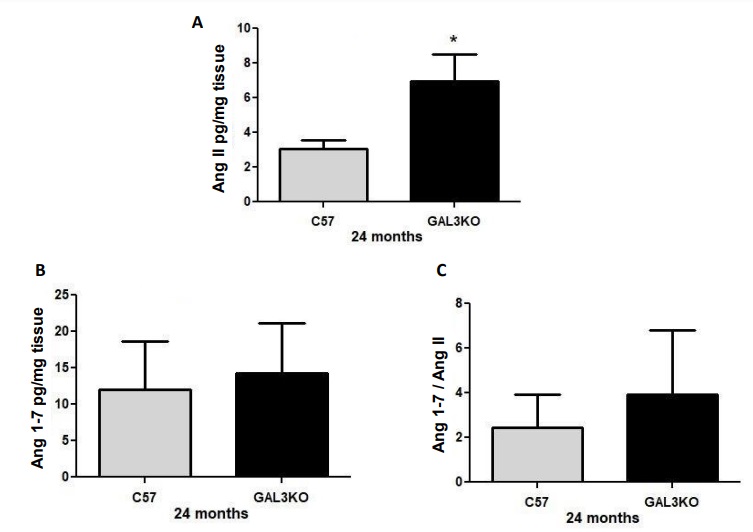
Increased myocardial expression of Ang II in aged Gal-3 KO mice
Cardiac expression of Ang II was significantly increased in old Gal-3 KO mice compared with control mice (Fig. 6A; p < 0.02). Cardiac Ang (1–7) and Ang 1-7/Ang II expression were similar between genotypes (Fig. 6B and C; p=NS).
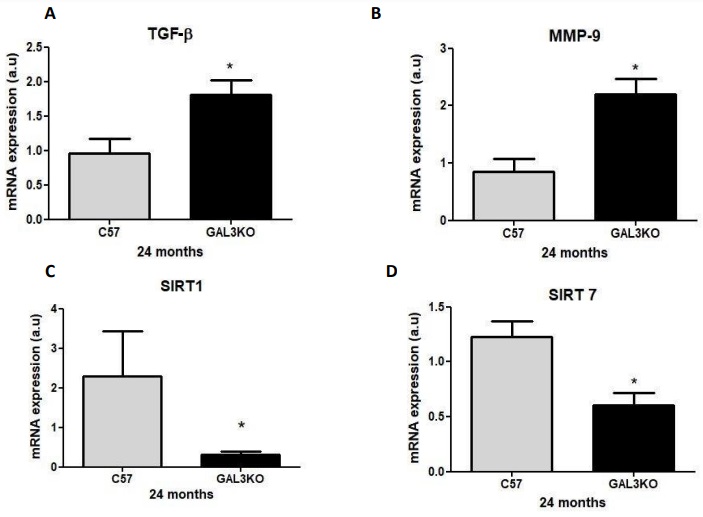
Increased cardiac expression of TGF- β and MMP-9 and reduced SIRT1 and 7 in aged Gal-3
KO mice
Cardiac expression of SIRT1 and 7 were significantly lower in aged Gal-3 KO mice than in control mice (Fig. 7C–D); p < 0.05). On the contrary, cardiac mRNA expression for the fibrotic factor TGF-β and the hypertrophy promotor MMP-9 were significantly increased in aged GAL-3 KO mice compared with C57 mice (Fig. 7A–B); p < 0.05).
The authors gratefully appreciate the technical assistance of Ana Chiaro, María Agustina Vidal, and Sergio Mazzini.
Author Contributions
FSFS, MCB, VM, IMS, VT, CS, AV, and GEG: Conceived and designed the protocol, performed the experiments, collected the data, performed the analysis, and wrote the paper. MS and MMG: processed the hearts, and performed the RIA and data analysis. FP and NG: processed the hearts, and performed and analyzed the RTq-PCR data as well as wrote the paper. TFC contributed to perform and analyzed the echocardiogram data; VM and CM performed the analysis of histopathological samples.
Funding
This work was supported by grants from the Argentinean Agency for Promotion of Science and Technology (PICT 2014-2320, 2019-02987 and PICTO 2017-055 to GEG, and PICT 2018-03267 to VM) and the University of Buenos Aires (UBACyT 2018-20020170100619BA to GEG).
Statement of Ethics
The experimental protocol conforms to internationally accepted standards and it was approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL) of BIOMED in line with the NIH’s Guide for the Care and Use of Laboratory Animals.
The authors declare that no conflicts of interest exist.
| 1 Damluji AA, Forman DE, van Diepen S, Alexander KP, Page RL, Hummel SL, Menon V, Katz JN, Albert NM, Afilalo J, Cohen MG: Older Adults in the Cardiac Intensive Care Unit: Factoring Geriatric Syndromes in the Management, Prognosis, and Process of Care: A Scientific Statement From the American Heart Association. Circulation 2020;141:e6-e32. https://doi.org/10.1161/CIR.0000000000000741 |
||||
| 2 Shock NW: Physiologic aspects of aging. J Am Diet Assoc 1970;56:491-496. https://doi.org/10.1016/S0002-8223(21)13351-6 |
||||
| 3 Harman D: Free radical theory of aging: Consequences of mitochondrial aging. AGE 1983;6:86-94. https://doi.org/10.1007/BF02432509 |
||||
| 4 Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS: Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxidants Redox Signal 2012;16:1492-1526. https://doi.org/10.1089/ars.2011.4179 |
||||
| 5 Keller KM, Howlett SE: Sex Differences in the Biology and Pathology of the Aging Heart. Can J Cardiol 2016;32:1065-1073. https://doi.org/10.1016/j.cjca.2016.03.017 |
||||
| 6 Guo L, Yin A, Zhang Q, Zhong T, O'Rourke ST, Sun C: Angiotensin-(1-7) attenuates angiotensin II-induced cardiac hypertrophy via a Sirt3-dependent mechanism. Am J Physiol Heart Circ Physiol 2017;312:H980-H991. https://doi.org/10.1152/ajpheart.00768.2016 |
||||
| 7 Nozato S, Yamamoto K, Takeshita H, Nozato Y, Imaizumi Y, Fujimoto T, Yokoyama S, Nagasawa M, Takeda M, Hongyo K, Akasaka H, Takami Y, Takeya Y, Sugimoto K, Mogi M, Horiuchi M, Rakugi H: Angiotensin 1-7 alleviates aging-associated muscle weakness and bone loss, but is not associated with accelerated aging in ACE2-knockout mice. Clin Sci (Lond) 2019;133:2005-2018. https://doi.org/10.1042/CS20190573 |
||||
| 8 Basso N, Paglia N, Stella I, De Cavanagh EMV, Ferder L, Arnaiz MDRL, Inserra F: Protective effect of the inhibition of the renin-angiotensin system on aging. Regul Pept 2005;128:247-252. https://doi.org/10.1016/j.regpep.2004.12.027 |
||||
| 9 Dai DF, Rabinovitch PS: Cardiac Aging in Mice and Humans: The Role of Mitochondrial Oxidative Stress. Trends Cardiovasc Med 2009;19:213-220. https://doi.org/10.1016/j.tcm.2009.12.004 |
||||
| 10 Horn MA, Trafford AW: Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J Mol Cell Cardiol 2016;93:175-185. https://doi.org/10.1016/j.yjmcc.2015.11.005 |
||||
| 11 Hsu CP, Odewale I, Alcendor RR, Sadoshima J: Sirt1 protects the heart from aging and stress. Biol Chem 2008;389:221-231. https://doi.org/10.1515/BC.2008.032 |
||||
| 12 Matsushima S, Sadoshima J: The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol 2015;309:H1375-H1389. https://doi.org/10.1152/ajpheart.00053.2015 |
||||
| 13 Toba H, Cannon PL, Yabluchanskiy A, Iyer RP, D'Armiento J, Lindsey ML: Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am J Physiol Heart Circ Physiol 2017;312:H375-H383. https://doi.org/10.1152/ajpheart.00633.2016 |
||||
| 14 Suthahar N, Meijers WC, Silljé HHW, Ho JE, Liu FT, de Boer RA: Galectin-3 activation and inhibition in heart failure and cardiovascular disease: An update. Theranostics 2018;8:593-609. https://doi.org/10.7150/thno.22196 |
||||
| 15 De Boer RA, Lok DJA, Jaarsma T, Van Der Meer P, Voors AA, Hillege HL, Van Veldhuisen DJ: Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann Med 2011;43:60-68. https://doi.org/10.3109/07853890.2010.538080 |
||||
| 16 Liu FT, Rabinovich GA: Galectins: Regulators of acute and chronic inflammation. Ann N Y Acad Sci 2010;1183:158-182. https://doi.org/10.1111/j.1749-6632.2009.05131.x |
||||
| 17 Sharma UC, Pokharel S, Van Brakel TJ, Van Berlo JH, Cleutjens JPM, Schroen B, André S, Crijns HJGM, Gabius HJ, Maessen J, Pinto YM: Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004;110:3121-3128. https://doi.org/10.1161/01.CIR.0000147181.65298.4D |
||||
| 18 Cassaglia P, Penas F, Betazza C, Fontana Estevez FS, Miksztowicz V, Martínez Naya N, Llamosas MC, Noli Truant S, Wilensky L, Volberg V, Cevey ÁC, Touceda V, Cicale E, Berg G, Fernández M, Goren N, Morales C, González GE: Genetic Deletion of Galectin-3 Alters the Temporal Evolution of Macrophage Infiltration and Healing Affecting the Cardiac Remodeling and Function after Myocardial Infarction in Mice. Am J Pathol 2020;190:1789-1800. https://doi.org/10.1016/j.ajpath.2020.05.010 |
||||
| 19 González GE, Rhaleb NE, D'Ambrosio MA, Nakagawa P, Liao TD, Peterson EL, Leung P, Dai X, Janic B, Liu YH, Yang XP, Carretero OA: Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 2016;311:H1287-H1296. https://doi.org/10.1152/ajpheart.00096.2016 |
||||
| 20 Yang RY, Rabinovich GA, Liu FT: Galectins: Structure, function and therapeutic potential. Expert Rev Mol Med 2008;10:e17. https://doi.org/10.1017/S1462399408000719 |
||||
| 21 de Boer RA, van Veldhuisen DJ, Gansevoort RT, Muller Kobold AC, van Gilst WH, Hillege HL, Bakker SJL, van der Harst P: The fibrosis marker galectin-3 and outcome in the general population. J Intern Med 2012;272:55-64. https://doi.org/10.1111/j.1365-2796.2011.02476.x |
||||
| 22 Díaz-Alvarez L, Soto E: The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediators Inflamm 2017;2017:9247574. https://doi.org/10.1155/2017/9247574 |
||||
| 23 Komici K, Gnemmi I, Bencivenga L, Vitale DF, Rengo G, Di Stefano A, Eleuteri E: Impact of galectin-3 circulating levels on frailty in elderly patients with systolic heart failure. J Clin Med 2020;9:1-12. https://doi.org/10.3390/jcm9072229 |
||||
| 24 Dong R, Zhang M, Hu Q, Zheng S, Soh A, Zheng Y, Yuan H: Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int J Mol Med 2018;41:599-614. https://doi.org/10.3892/ijmm.2017.3311 |
||||
| 25 Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, Larson MG, Levy D: Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 2012;60:1249-1256. https://doi.org/10.1016/j.jacc.2012.04.053 |
||||
| 26 Iacobini C, Oddi G, Menini S, Amadio L, Ricci C, Di Pippo C, Sorcini M, Pricci F, Pugliese F, Pugliese G: Development of age-dependent glomerular lesions in galectin-3/AGE-receptor- 3 knockout mice. Am J Physiol Ren Physiol 2005;289:F611-F621. https://doi.org/10.1152/ajprenal.00435.2004 |
||||
| 27 Albus U: Guide for the Care and Use of Laboratory Animals (8th ed). Lab Anim 2012; DOI: 10.1258/la.2012.150312. https://doi.org/10.1258/la.2012.150312 |
||||
| 28 Silva MG, Falcoff NL, Corradi GR, Alfie J, Seguel RF, Tabaj GC, Iglesias LI, Nuñez M, Guman GR, Gironacci MM: Renin-angiotensin system blockade on angiotensin-converting enzyme 2 and TMPRSS2 in human type II pneumocytes. Life Sci 2022;293:120324. https://doi.org/10.1016/j.lfs.2022.120324 |
||||
| 29 Schmittgen TD, Livak KJ: Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3:1101-1108. https://doi.org/10.1038/nprot.2008.73 |
||||
| 30 Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT: The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009;55:611-622. https://doi.org/10.1373/clinchem.2008.112797 |
||||
| 31 Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J: Aging and Autophagy in the Heart. Circ Res 2016;118:1563-1576. https://doi.org/10.1161/CIRCRESAHA.116.307474 |
||||
| 32 Wang M, Zhao D, Spinetti G, Zhang J, Jiang LQ, Pintus G, Monticone R, Lakatta EG: Matrix Metalloproteinase 2 Activation of Transforming Growth Factor-β1 (TGF-β1) and TGF-β1-Type II Receptor Signaling Within the Aged Arterial Wall. Arterioscler Thromb Vasc Biol 2006;26:1503-1509. https://doi.org/10.1161/01.ATV.0000225777.58488.f2 |
||||
| 33 Loffredo FS, Nikolova AP, Pancoast JR, Lee RT: Heart Failure With Preserved Ejection Fraction. Circ Res 2014;115:97-107. https://doi.org/10.1161/CIRCRESAHA.115.302929 |
||||
| 34 Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML: The impact of aging on cardiac extracellular matrix. GeroScience 2017;39:7-18. https://doi.org/10.1007/s11357-017-9959-9 |
||||
| 35 Kasacka I, Piotrowska Z, Niezgoda M, Lewandowska A, Łebkowski W: Ageing-related changes in the levels of β-catenin, CacyBP/SIP, galectin-3 and immunoproteasome subunit LMP7 in the heart of men. PLoS One 2020;15:e0229462. https://doi.org/10.1371/journal.pone.0229462 |
||||
| 36 Nangia-Makker P, Nakahara S, Hogan V, Raz A: Galectin-3 in apoptosis, a novel therapeutic target. J Bioenerg Biomembr 2007;39:79-84. https://doi.org/10.1007/s10863-006-9063-9 |
||||
| 37 Conti S, Cassis P, Benigni A: Aging and the renin-angiotensin system. Hypertension 2012;60:878-883. https://doi.org/10.1161/HYPERTENSIONAHA.110.155895 |
||||
| 38 Yoon HE, Kim EN, Kim MY, Lim JH, Jang IA, Ban TH, Shin SJ, Park CW, Chang YS, Choi BS: Age-Associated Changes in the Vascular Renin-Angiotensin System in Mice. Oxid Med Cell Longev 2016;2016:6731093. https://doi.org/10.1155/2016/6731093 |
||||
| 39 Dostal DE, Rothblum KN, Chernin MI, Cooper GR, Baker KM: Intracardiac detection of angiotensinogen and renin: a localized renin-angiotensin system in neonatal rat heart. Am J Physiol1992;263:C838-C850. https://doi.org/10.1152/ajpcell.1992.263.4.C838 |
||||
| 40 Raman VK, Lee YA, Lindpaintner K: The cardiac renin-angiotensin-aldosterone system and hypertensive cardiac hypertrophy. Am J Cardiol 1995;76:18D-23D. https://doi.org/10.1016/S0002-9149(99)80487-1 |
||||
| 41 Varagic J, Frohlich ED: Local cardiac renin-angiotensin system: hypertension and cardiac failure. J Mol Cell Cardiol 2002;34:1435-1442. https://doi.org/10.1006/jmcc.2002.2075 |
||||
| 42 Sadoshima J, Xu Y, Slayter HS, Izumo S: Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro . Cell 1993;75:977-984. https://doi.org/10.1016/0092-8674(93)90541-W |
||||
| 43 Saupe KW, Sobol SC, Koh SG, Apstein CS: Effects of AT1 receptor block begun late in life on normal cardiac aging in rats. J Cardiovasc Pharmacol 2003;42:573-580. https://doi.org/10.1097/00005344-200310000-00017 |
||||
| 44 Miller AJ, Bingaman SS, Mehay D, Medina D, Arnold AC: Angiotensin-(1-7) Improves Integrated Cardiometabolic Function in Aged Mice. Int J Mol Sci 2020;21:1-13. https://doi.org/10.3390/ijms21145131 |
||||
| 45 Varagic J, Ahmad S, Nagata S, Ferrario CM: ACE2: angiotensin II/angiotensin-(1-7) balance in cardiac and renal injury. Curr Hypertens Rep 2014;16:420. https://doi.org/10.1007/s11906-014-0420-5 |
||||
| 46 Al-Salam S, Hashmi S, Jagadeesh GS, Tariq S: Galectin-3: A Cardiomyocyte Antiapoptotic Mediator at 24-Hour Post Myocardial Infarction. Cell Physiol Biochem 2020;54:287-302. https://doi.org/10.33594/000000220 |
||||
| 47 Sun Z, Zhang L, Li L, Shao C, Liu J, Zhou M, Wang Z: Galectin-3 mediates cardiac remodeling caused by impaired glucose and lipid metabolism through inhibiting two pathways of activating Akt. Am J Physiol Heart Circ Physiol 2021;320:H364-H380. https://doi.org/10.1152/ajpheart.00523.2020 |
||||
| 48 Li X, Tang X, Lu J, Yuan S: Therapeutic inhibition of galectin‑3 improves cardiomyocyte apoptosis and survival during heart failure. Mol Med Rep 2018;17:4106-4112. https://doi.org/10.3892/mmr.2017.8323 |
||||
| 49 Zhang M, Cheng K, Chen H, Tu J, Shen Y, Pang L, Wu W: Galectin-3 knock down inhibits cardiac ischemia-reperfusion injury through interacting with bcl-2 and modulating cell apoptosis. Arch Biochem Biophys 2020;694:108602. https://doi.org/10.1016/j.abb.2020.108602 |
||||
| 50 Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C: Functional Role of Phosphodiesterase 3 in Cardiomyocyte Apoptosis Implication in Heart Failure. Circulation 2005;111:2469-2476. https://doi.org/10.1161/01.CIR.0000165128.39715.87 |
||||
| 51 Hanna A, Frangogiannis NG: The Role of the TGF-β Superfamily in Myocardial Infarction. Front Cardiovasc Med 2019;6:140. https://doi.org/10.3389/fcvm.2019.00140 |
||||
| 52 Suzuki M, Ramezanpour M, Cooksley C, Li J, Nakamaru Y, Homma A, Psaltis A, Wormald PJ, Vreugde S: Sirtuin-1 Controls Poly (I:C)-Dependent Matrix Metalloproteinase 9 Activation in Primary Human Nasal Epithelial Cells. Am J Respir Cell Mol Biol 2018;59:500-510. https://doi.org/10.1165/rcmb.2017-0415OC |
||||
| 53 Liu P, Su J, Song X, Wang S: miR-92a regulates the expression levels of matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 3 via sirtuin 1 signaling in hydrogen peroxide-induced vascular smooth muscle cells. Mol Med Rep 2018;17:1041-1048. https://doi.org/10.3892/mmr.2017.7937 |
||||