×
![]()
Corresponding Author: Pin-Lan Li
Department of Pharmacology & Toxicology, Medical College of Virginia Campus, Virginia Commonwealth University, Richmond, VA 23298 (USA)
Tel. +1 (804) 828-4793, Fax +1 (804) 828-4794 , E-Mail pin-lan.li@vcuhealth.org
Lysosome Function in Cardiovascular Diseases
Owais M. Bhat Pin-Lan Li
Department of Pharmacology and Toxicology, Virginia Commonwealth University, School of Medicine, Richmond, VA, USA
Introduction
Christian de Duve, in 1974 received the Nobel Prize for his work on lysosome structure and functions. Lysosomes are acidic, spherical, membrane-bound organelles within a cell. They contain various hydrolytic enzymes that catalyze hydrolysis reactions. The synthesis of lysosome proteins is similar to other proteins. Hydrolytic enzymes synthesized in rough endoplasmic reticulum and tagged with mannose-6-phosphate within the Golgi apparatus are targeted to the lysosome. Vesicles containing these hydrolytic enzymes bud off from the Golgi apparatus and in the cytoplasm, these vesicles bind with late endosomes. The late endosomes can eventually mature into lysosomes. H+-ATPase residing on lysosomal membrane leads to acidification of lysosomes, which facilitates the activity of various acid hydrolases. Entry of calcium (Ca2+) into lysosomal compartment is carried out by H+/Ca2+ exchange under resting condition and released under various stimulations [1, 2].
The hydrolytic enzymes within the lysosome allow them to destroy foreign particles via a cellular process known as phagocytosis. Lysosomes provide a defense system to the cell against entry of various pathogens via endocytic process before these pathogens are delivered to the cytoplasm [3]. The enzymes within the lysosomes work in an oxygen-independent mechanism in killing various pathogens. In addition, the lysosomes are involved in breakdown of many biomolecules, misfolded proteins and damaged organelles as part of the recycling system of the cell [4]. Moreover, lysosomes also play an important role in oocyte maturation and fertilization during acrosome reaction, the sperm head contains lysosomal enzymes which effectively bore a hole through the egg membrane, thereby facilitating the entry of sperm into the egg [5].
In addition to its crucial role in phagocytosis, the lysosome has been well known to participate in autophagy, a catabolic process to degrade cytoplasmic components and organelles that maintain cellular homeostasis. The term “Autophagy” term was coined in the 1960s and was classified into three categories, which include microautophagy, macroautophagy and chaperone-mediated autophagy. Macroautophagy is a major regulator of catabolic mechanisms and has been well characterized in eukaryotic cells, this process is used to degrade damaged or long-lived proteins and organelles [6, 7]. Although the autophagy term was coined in 1960s, knowledge regarding its morphological and biochemical characteristics was unveiled in the early 1990s. In the past decade, owing to the discovery of yeast autophagy genes (Atg genes) followed by their identification with the mammalian homologues, several studies elucidated the molecular machinery of this main cellular homeostatic process and its regulatory mechanisms [8, 9]. The autophagic process involves 4 stages including induction, autophagosome (AP) formation, docking and fusion with lysosomes (namely, the formation of autophagolysosome (APLs) or autolysosomes), and breakdown of autophagic vesicles [10]. Our laboratory also focused on such mechanisms revealing that the normal regulation of lysosome trafficking and fusion is controlled by nicotinic acid adenine dinucleotide phosphate (NAADP) or ceramide as well as lysosomal and cytosolic Ca2+ levels [11].
Lysosomal storage disorders (LSDs) are major category of lysosome dysfunction that contributes to cardiovascular disorders. Deficiency of lysosomal enzymes, membrane transporters, or several other proteins that are involved in lysosomal biology are main causes of LSDs [12]. Many patients suffering from LSDs show severe cardiac phenotypes including coronary artery disorders. Mutational disorders in lysosomal genes have been identified as causative factors, which are responsible for the disease pathogenesis. For example, Fabry disease is caused by a deficiency in the lysosomal enzyme alpha-galactosidase A (α-Gal A), an enzyme involved in sphingolipid metabolism, leading to buildup of the fatty acid globotriaosylceramide (Gb3) in the walls of the blood vessels and other organs of the body [13]. Besides LSDs, lysosome dysfunction has been recently reported to play an important role in the development of different human diseases [14, 15]. This review will briefly summarize current evidences that lysosome regulation and dysfunction may be implicated in the pathogenesis or pathophysiology of cardiovascular diseases such as vascular calcification, arterial stiffening, and atherosclerosis.
Lysosome and Cardiovascular Diseases
With respect to cardiovascular regulation and disease, many studies have demonstrated that abnormal autophagy including autophagic flux has a variety of pathogenic actions on the cardiovascular system. For example, Transcription Factor EB (TFEB) is a transcription factor, master regulator of autophagy and lysosome biogenesis genes. Macrophage-specific overexpression of TFEB in a mice model lead to atheroprotection. It was observed that overexpression of TFEB is associated with atheroprotection including reductions in plaque burden including apoptotic and necrotic areas [16]. Mechanistically, TFEB decreases accumulation of ubiquitinated and SQSTM1-enriched protein aggregates, IL1B/IL-1β levels, and macrophage apoptosis. TFEB stimulates endocytosis, phagocytosis that help macrophages to engulf apoptotic cells in atherosclerosis. In addition, TFEB drives the expression of lipid metabolic and mitochondrial genes via transcriptional activation of PPARGC1A/PGC-1α. Deficiency of lysosomal-associated membrane protein-2 (Lamp-2) gene, which encodes for a lysosomal membrane protein on chromosome X causes Danon disease, which often leads to cardiomyopathy/ heart failure. In human cardiomyocytes, autophagosome-lysosome fusion requires Lamp-2 isoform B (Lamp-2B). In addition, gene correction of Lamp-2 mutation rescues the Danon phenotype [17]. This study provided an evidence for cardiomyopathy in Danon patients and suggested defective Lamp-2B–mediated autophagy as a therapeutic target to treat this patient population.
Lysosome has been known to regulate endothelial function, and it participates in transmembrane signaling of different death receptors via formation of membrane rafts (MRs) redox signaling platforms, thereby leading to endothelial dysfunction upon different stimuli [18]. During atherosclerosis, lysosome-associated membrane signalosome plays a crucial role in endothelial injury such as abnormal leukocyte adhesion, invasion or infiltration of macrophages, and local oxidative stress. During atherosclerosis, macrophages uptake the oxidized form of cholesterol through scavenger receptors and deliver to lysosomes through endocytic process for degradation. Under normal condition, lysosomal acid lipase hydrolyzed the cholesteryl esters into free cholesterol, which is then transported in an ATP dependent process out of lysosomes through lysosomal Niemann-Pick type C1 protein. This catabolism and transport of cholesterol in lysosomes are regulated by a number of lysosomal molecules such as acid sphingomyelinase (ASMase), mucolipin-1, and H+-ATPase. Any defect in these events may cause accumulation of cholesterol into the lysosomes and deficient clearance of cholesterol from macrophages. These events lead to lipid deposition, foam cell formation and ultimately to atherosclerosis. Cardiovascular disease is the leading cause of death worldwide. According to the American Heart Association 2018 report [19], cardiovascular disease accounts for more than 800,000, or approximately one in three, deaths in the United States each year. Although, there are various underlying causes or causative factors which contribute to the pathogenesis of cardiovascular diseases, lysosome function is strongly correlated with the development and progression of these diseases [15]. More detailed information about the critical role of lysosomes in several major vascular diseases is discussed below.
Vascular Calcification
Vascular calcification is a pathology characterized by deposition of dispersed punctate or hydroxyapatite patchy crystals. It is characteristic of aging and also contributes to diabetes mellitus, atherosclerosis and chronic kidney disease (CKD) [20]. Vascular calcification localized to atherosclerotic neointima is known as intimal calcification and is detected as microcalcification (range: ≥0.5 to <15 μm). It is assumed that microcalcification is originated from apoptotic smooth muscle cells (SMC) or matrix vesicles that are released by these SMCs. It occurs near the internal elastic lamina and is associated with lipid deposition and inflammation in the neointima [21]. Vascular calcification histologically located in medial layer of the vessel, known as arterial medial calcification (AMC), surrounding the arterial medial SMCs and along the elastic lamellae is also known as Monckeberg’s medial sclerosis. It reduces the arterial compliance and is prevalent in diabetes mellitus and CKD. The pathogenesis of AMC is still poorly understood however; the process is believed to mimic the skeletal bone formation [22]. AMC is a complex and highly regulated process which involves the reprogramming and osteochondrogenic differentiation of vascular smooth muscle cells (VSMCs) and secretion of calcifying matrix vesicles or apoptotic bodies generated from these VSMCs which initiates deposition of calcium/phosphate (Ca2+/ Pi) crystals in the arterial wall [23-25].
From the last decade, studies are citing the role of sphingolipids in vascular calcification. A study in VSMCs showed that sphingosine-1-phosphate (S1P) stimulates the phosphorylation of ezrin-radixinmoesin (ERM) axis increasing mineralization; however, inhibition of ASMase and ceramidase prevented S1P level increase, ERM activation, and mineralization [26]. S1P is also involved in trans-differentiation and calcification of valve interstitial cells that contributes to valve calcification [27]. Song et al. [28] showed that TLR4/NF-κB/Ceramide signaling mediates Ox-LDL-induced calcification of human VSMCs. In patients with cystic echinococcosis (CE), relative expression of Asah1 gene (codes for acid ceramidase) was low in patients with calcification [29]. In human femoral arterial SMCs, Ox-LDL-induced matrix mineralization was mediated via ceramide, which was attributed to increased neutral-sphingomyelinase (N-SMase) activity and ceramide levels [30]. Kapustin et al. also reported that N-SMase2 inhibition reduces mineralization in response to osteogenic medium in human coronary artery SMCs [25]. In sphingolipid catabolism, markedly increased levels of Sphingosine-1-Phosphate Lyase 1 (SGPL1) substrates, S1P and sphingosine was observed in the patient’s blood and fibroblasts, accompanied with adrenal calcifications and vascular alterations in renal biopsies, which were consistent with changes seen in Sgpl1 knockout mice [31]. A study in patients with coronary calcification identified 103 lipids including the sphingolipid and sterol lipid classes might aid in better assessment of patients with subclinical coronary artery disease [32].
Recently, our group explored the role of lysosomal-sphingolipid metabolism in the vascular calcification and these findings provide the first experimental evidence in this area. Our findings revealed that SMC specific deletion of acid ceramidase (Ac) or overexpression of ASMase leads to the accumulation of ceramide in the arterial SMCs which plays a key role in osteogenic phenotype transition, increased small extracellular vesicle (sEV) secretion and mineral deposition in these cells that contribute to AMC. In addition, we found that GW4869, a N-SMase inhibitor significantly decreased Pi-induced calcification in coronary arterial smooth muscle cells (CASMCs) [33]. Moreover, ASMase inhibition by amitriptyline, a pharmacological inhibitor of ASMase significantly reduced CASMCs calcification both in vitro and in vivo [34]. These findings provide a novel insight into the molecular mechanisms associated with the sphingolipid-ceramide pathway required for osteogenic lineage reprogramming of SMCs that result in AMC, and indicate new therapeutic strategies for the prevention and treatment of vascular calcification. Three major molecular mechanisms as discussed below are proposed to address the contribution of lysosomes and related enzymes to the vascular calcification.
Phenotypic change of VSMCs. VSMCs can undergo phenotypic switching from contractile (differentiated) phenotype to synthetic (dedifferentiated) phenotype in response to various stimuli including growth factors, cell adhesion molecules, chemotactic factors, extracellular matrix (ECM) enzymes, and injury stimuli signals [35, 36]. This phenotypic transition of VSMCs is associated with their proliferation and is one of the major contributing factors for the initiation of vascular remodeling in hypertension, atherosclerosis and vascular restenosis [37, 38]. Synthetic or dedifferentiated VSMCs showed increased viability in proliferation, migration, and synthesis, and reduced expression of differentiation markers α-SMA and SM22-α [39, 40].
VSMCs have been shown to undergo differentiation to osteoblast-like cells. Bone-related transcription factors, including MSX2, RUNX2, SOX9, and osterix, which promote osteogenesis have been found in SMCs in calcified blood vessels. Osteo inductive cytokines such as tumor necrosis factor-α (TNF- α) upregulate expression of the transcription factors RUNX2 and osterix via activation of MSX2 and Wnt signaling pathway [22, 41]. In high-fat diet fed low-density lipoprotein (LDL) receptor knockout mouse model, increased serum TNF-α levels were found to be associated with augmented aortic expression of bone morphogenetic protein-2 (BMP-2), MSX2, Wnt3a, Wnt7a and aortic calcification [42]. RUNX2, a protein related to osteoblast differentiation in turn upregulates various bone-related proteins such as osteocalcin, sclerostin, and receptor activator of nuclear factor-kappaβ ligand (RANKL) [43]. Osterix activated a repertoire of genes during differentiation of preosteoblasts into mature osteoblasts and osteocytes including bone sialoprotein and alkaline phosphatase (ALP) [22, 44, 45].
Under normal physiological conditions, spontaneous accumulation of Ca2+/Pi levels are tightly balanced in the vasculature [46]. However, imbalanced mineral metabolism led to increased intracellular phosphate levels in VSMCs which directly drive their osteogenic differentiation and mineralization, inducing expression of osteogenic markers as shown in Fig. 1 [47, 48]. Under in vitro conditions, VSMCs exposed to a calcifying environment lost SMC lineage markers such as SM22α and SM α-actin and increased expression of the osteogenic markers such as RUNX2, osteocalcin, osteopontin, and ALP was observed [47]. In addition, increased expression of RUNX2 independent of downregulation of myocardin and SMC contractile proteins was found to be important for osteogenic switch and calcification [49]. Ex vivo human samples and animal models of arterial calcification demonstrated that free serum Ca2+/Pi levels caused ossification of soft tissue [50-52]. Increased serum Ca2+/Pi levels are correlated with the development and progression of calcification in human subjects [53]. Mineral imbalance actively stimulates phenotypic transformation of VSMCs during calcification process. In vitamin D (Vit D)-induced calcification mouse model, increased RUNX2 expression was observed [54], akin to CKD patient’s high doses of Vit D is correlated with severity of calcification [55]. Several in vivo studies demonstrated that physiologically Vit D promotes AMC through abnormal mineral metabolism (Ca2+/Pi), which as reported lead to vascular osteogenesis and mineralization [56, 57]. Our study in Smpd1trg/SMcre mice with SMC‐specific overexpression of Smpd1 gene showed that lysosomal acid sphingomyelinase (murine gene code: Smpd1)-derived ceramide contributes to the phenotypic switch in SMCs which leads to AMC. A high dose of Vit D (500000 IU/kg/d) resulted in increased AMC associated with augmented expression of RUNX2 and osteopontin in the coronary and aortic media, indicating phenotypic switch. However, amitriptyline, an ASMase inhibitor, reduced calcification and reversed phenotypic switch. These data indicate that lysosomal ceramide plays a critical role in phenotype change in SMCs, which may contribute to the arterial stiffness during the development of AMC [34]. Furthermore, our group reported that Asah1 gene (which encodes for acid ceramidase (Ac)) deletion specifically in SMCs in Asah1fl/fl/SMcre mice displayed more severe AMC in both coronary arteries and aorta receiving a high dose of Vit D which contributed to phenotypic change in arterial medial SMCs. Marked increase in osteopontin and RUNX2 (osteogenic markers) was observed in the arterial media of these mice [33]. These findings were validated in vitro using cultured CASMCs from Asah1fl/fl/SMcre mice treated with high Pi. In this study, we also found that Lysosomal transient receptor potential mucolipin 1 (TRPML1) channels regulating lysosome interaction with multivesicular bodies (MVBs) contributes to this phenotypic switch in these cells. Furthermore, in another study, we employed Vit D-induced mucolipin knockout mice model to explore the role of TRPML1 channel in the AMC. We found that lysosomal expression of mucolipin-1, a product of the mouse Mcoln1 gene, which regulates lysosomal positioning contributes to the phenotypic transition of arterial SMCs [58].
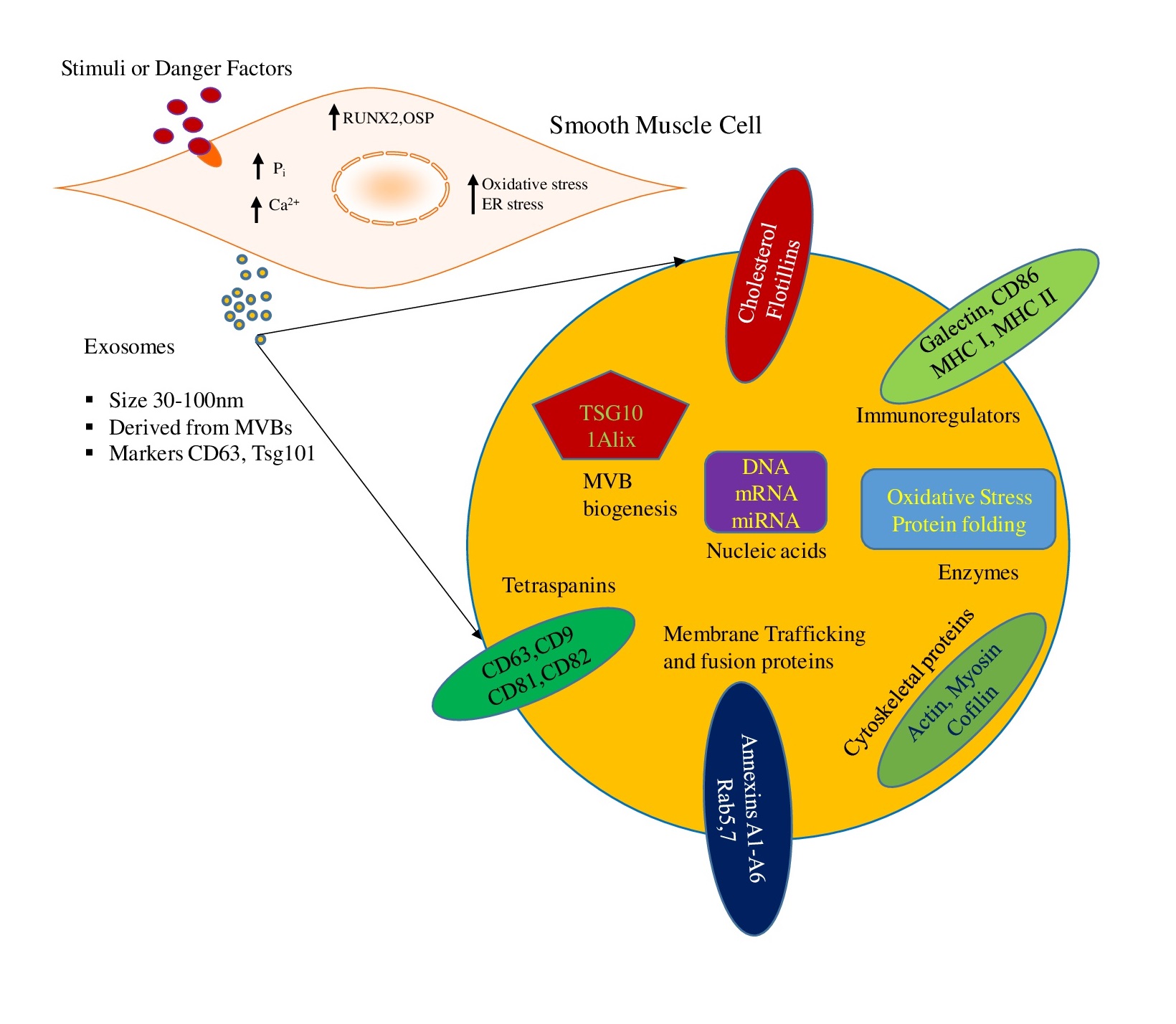
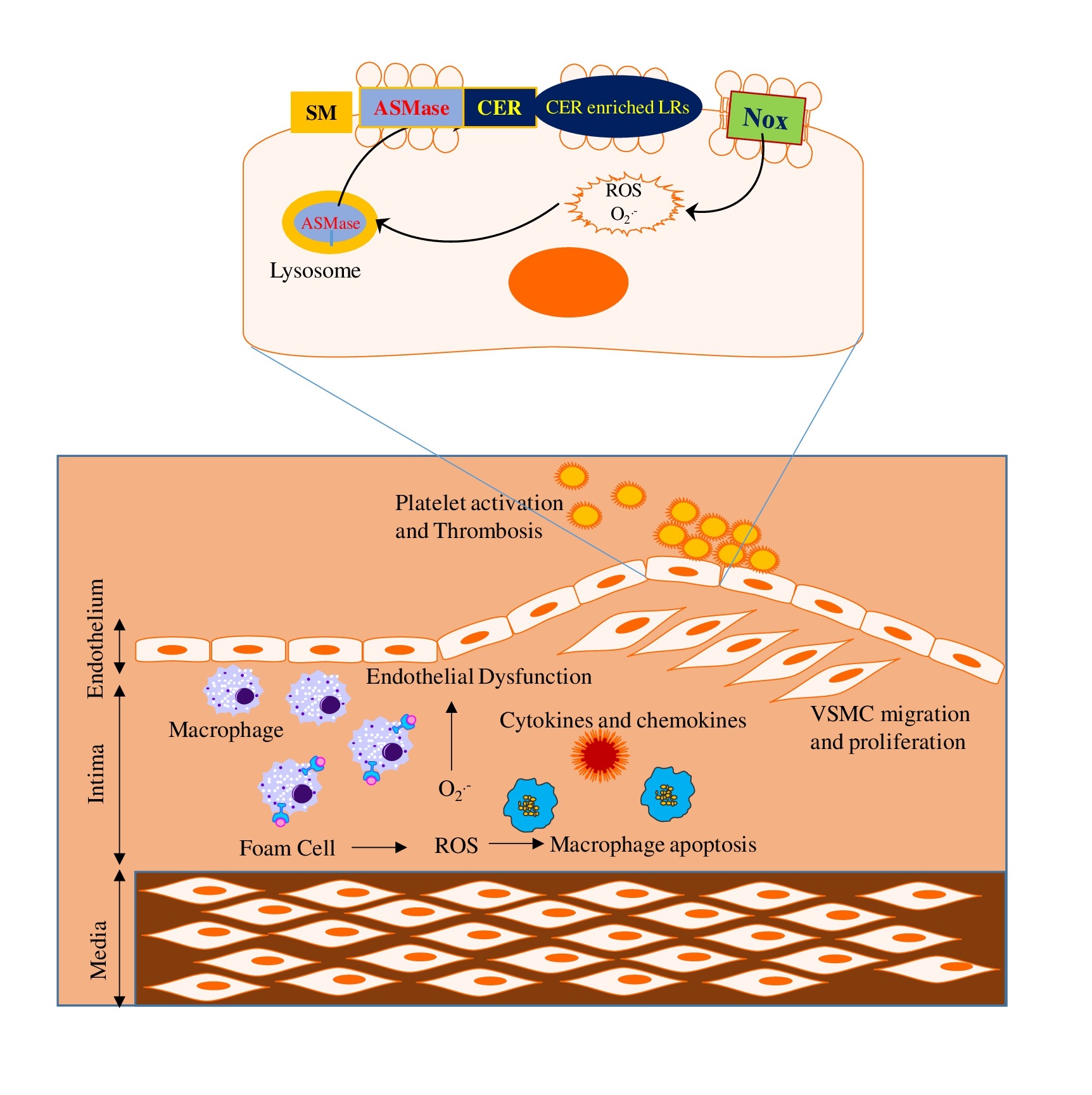
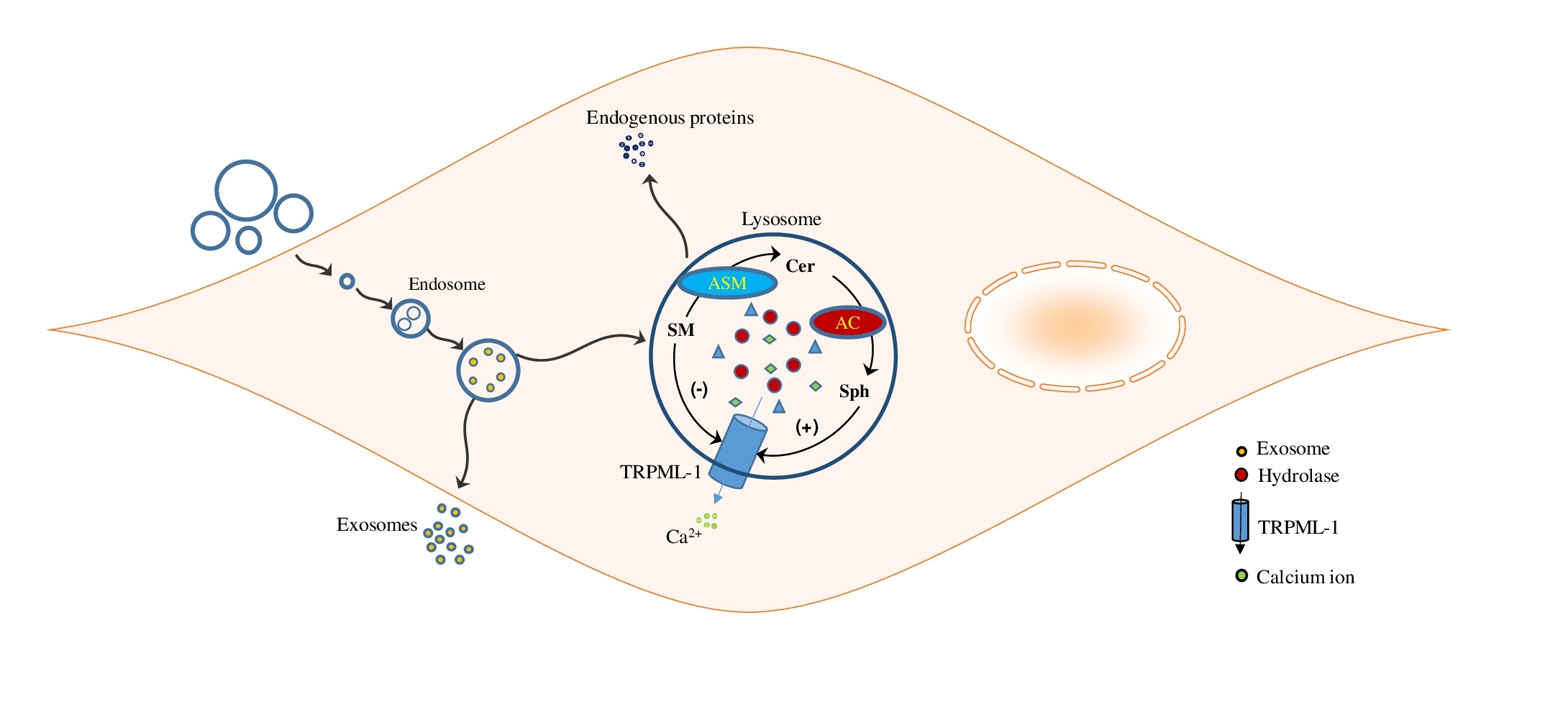
Most of the works cited in this review from authors’ laboratories were supported by grants from the National Institutes of Health (HL057244, HL075316 and DK120491).
The authors have no conflicts of interests to disclose.
| 1 Christensen KA, Myers JT, Swanson JA: pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci 2002;115:599-607. https://doi.org/10.1242/jcs.115.3.599 |
||||
| 2 Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A: NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002;111:703-708. https://doi.org/10.1016/S0092-8674(02)01082-6 |
||||
| 3 Lawrence RE, Zoncu R: The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 2019;21:133-142. https://doi.org/10.1038/s41556-018-0244-7 |
||||
| 4 Settembre C, Fraldi A, Medina DL, Ballabio A: Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013;14:283-296. https://doi.org/10.1038/nrm3565 |
||||
| 5 Jha KN, Kameshwari DB, Shivaji S: Role of signaling pathways in regulating the capacitation of mammalian spermatozoa. Cell Mol Biol (Noisy-le-grand) 2003;49:329-340. | ||||
| 6 Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC: Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010;90:1383-1435. https://doi.org/10.1152/physrev.00030.2009 |
||||
| 7 Levine B, Kroemer G: Autophagy in the pathogenesis of disease. Cell 2008;132:27-42. https://doi.org/10.1016/j.cell.2007.12.018 |
||||
| 8 Levine B, Klionsky DJ: Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004;6:463-477. https://doi.org/10.1016/S1534-5807(04)00099-1 |
||||
| 9 Wang W, Xu J, Kirsch T: Annexin-mediated Ca2+ influx regulates growth plate chondrocyte maturation and apoptosis. J Biol Chem 2003;278:3762-3769. https://doi.org/10.1074/jbc.M208868200 |
||||
| 10 Reggiori F, Klionsky DJ: Autophagy in the eukaryotic cell. Eukaryot Cell 2002;1:11-21. https://doi.org/10.1128/EC.01.1.11-21.2002 |
||||
| 11 Xu M, Zhang Y, Xia M, Li XX, Ritter JK, Zhang F, Li PL: NAD(P)H oxidase-dependent intracellular and extracellular O2*- production in coronary arterial myocytes from CD38 knockout mice. Free Radic Biol Med 2012;52:357-365. https://doi.org/10.1016/j.freeradbiomed.2011.10.485 |
||||
| 12 Winchester B, Vellodi A, Young E: The molecular basis of lysosomal storage diseases and their treatment. Biochem Soc Trans 2000;28:150-154. https://doi.org/10.1042/bst0280150 |
||||
| 13 Karen JK, Hale EK, Ma L: Angiokeratoma corporis diffusum (Fabry disease). Dermatol Online J 2005;11:8. | ||||
| 14 Ballabio A, Gieselmann V: Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 2009;1793:684-696. https://doi.org/10.1016/j.bbamcr.2008.12.001 |
||||
| 15 Wang X, Robbins J: Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol 2014;71:16-24. https://doi.org/10.1016/j.yjmcc.2013.11.006 |
||||
| 16 Evans TD, Jeong SJ, Zhang X, Sergin I, Razani B: TFEB and trehalose drive the macrophage autophagy-lysosome system to protect against atherosclerosis. Autophagy 2018;14:724-726. https://doi.org/10.1080/15548627.2018.1434373 |
||||
| 17 Chi C, Leonard A, Knight WE, Beussman KM, Zhao Y, Cao Y, Londono P, Aune E, Trembley MA, Small EM, Jeong MY, Walker LA, Xu H, Sniadecki NJ, Taylor MR, Buttrick PM, Song K: LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc Natl Acad Sci U S A 2019;116:556-565. https://doi.org/10.1073/pnas.1808618116 |
||||
| 18 Zhang AY, Yi F, Zhang G, Gulbins E, Li PL: Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006;47:74-80. https://doi.org/10.1161/01.HYP.0000196727.53300.62 |
||||
| 19 Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, et al.: Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018;137:e67-e492. https://doi.org/10.1161/CIR.0000000000000573 |
||||
| 20 Sage AP, Tintut Y, Demer LL: Regulatory mechanisms in vascular calcification. Nat Rev Cardiol 2010;7:528-536. https://doi.org/10.1038/nrcardio.2010.115 |
||||
| 21 Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R: Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol 2014;34:724-736. https://doi.org/10.1161/ATVBAHA.113.302642 |
||||
| 22 Bostrom KI, Rajamannan NM, Towler DA: The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res 2011;109:564-577. https://doi.org/10.1161/CIRCRESAHA.110.234278 |
||||
| 23 Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME: Medial localization of mineralization-regulating proteins in association with Monckeberg's sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation 1999;100:2168-2176. https://doi.org/10.1161/01.CIR.100.21.2168 |
||||
| 24 Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL: Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res 2000;87:1055-1062. https://doi.org/10.1161/01.RES.87.11.1055 |
||||
| 25 Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ, Shanahan CM: Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 2015;116:1312-1323. https://doi.org/10.1161/CIRCRESAHA.116.305012 |
||||
| 26 Morris TG, Borland SJ, Clarke CJ, Wilson C, Hannun YA, Ohanian V, Canfield AE, Ohanian J: Sphingosine 1-phosphate activation of ERM contributes to vascular calcification. J Lipid Res 2018;59:69-78. https://doi.org/10.1194/jlr.M079731 |
||||
| 27 Fernandez-Pisonero I, Lopez J, Onecha E, Duenas AI, Maeso P, Crespo MS, San Roman JA, Garcia-Rodriguez C: Synergy between sphingosine 1-phosphate and lipopolysaccharide signaling promotes an inflammatory, angiogenic and osteogenic response in human aortic valve interstitial cells. PLoS One 2014;9:e109081. https://doi.org/10.1371/journal.pone.0109081 |
||||
| 28 Son DJ, Jung YY, Seo YS, Park H, Lee DH, Kim S, Roh YS, Han SB, Yoon DY, Hong JT: Interleukin-32alpha Inhibits Endothelial Inflammation, Vascular Smooth Muscle Cell Activation, and Atherosclerosis by Upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis. Theranostics 2017;7:2186-2203. https://doi.org/10.7150/thno.18407 |
||||
| 29 Yin YJ, Zhang Q, Yang YX, Yang SK, Wang HF, Shi JX, Wang ZM, Yang YH, Lin Y, Li ZY, Yang YR: [Study of the genes correlated with cyst calcification in patients with cystic echinococcosis]. Zhonghua Yu Fang Yi Xue Za Zhi 2016;50:434-438. | ||||
| 30 Liao L, Zhou Q, Song Y, Wu W, Yu H, Wang S, Chen Y, Ye M, Lu L: Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS One 2013;8:e82379. https://doi.org/10.1371/journal.pone.0082379 |
||||
| 31 Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, Krainer I, Huber LA, Hess MW, Frishberg Y, Barash H, Tzur S, Schreyer-Shafir N, Sukenik-Halevy R, Zehavi T, Raas-Rothschild A, Mao C, Muller T: Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum Mutat 2017;38:365-372. https://doi.org/10.1002/humu.23192 |
||||
| 32 Djekic D, Pinto R, Repsilber D, Hyotylainen T, Henein M: Serum untargeted lipidomic profiling reveals dysfunction of phospholipid metabolism in subclinical coronary artery disease. Vasc Health Risk Manag 2019;15:123-135. https://doi.org/10.2147/VHRM.S202344 |
||||
| 33 Bhat OM, Li G, Yuan X, Huang D, Gulbins E, Kukreja RC, Li PL: Arterial Medial Calcification through Enhanced small Extracellular Vesicle Release in Smooth Muscle-Specific Asah1 Gene Knockout Mice. Sci Rep 2020;10:1645. https://doi.org/10.1038/s41598-020-58568-5 |
||||
| 34 Bhat OM, Yuan X, Cain C, Salloum FN, Li PL: Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: A ceramide-mediated vasculopathy. J Cell Mol Med 2020;24:539-553. https://doi.org/10.1111/jcmm.14761 |
||||
| 35 Yoshida T, Owens GK: Molecular determinants of vascular smooth muscle cell diversity. Circ Res 2005;96:280-291. https://doi.org/10.1161/01.RES.0000155951.62152.2e |
||||
| 36 Owens GK, Kumar MS, Wamhoff BR: Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84:767-801. https://doi.org/10.1152/physrev.00041.2003 |
||||
| 37 Chistiakov DA, Orekhov AN, Bobryshev YV: Vascular smooth muscle cell in atherosclerosis. Acta Physiol (Oxf) 2015;214:33-50. https://doi.org/10.1111/apha.12466 |
||||
| 38 Bennett MR, Sinha S, Owens GK: Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res 2016;118:692-702. https://doi.org/10.1161/CIRCRESAHA.115.306361 |
||||
| 39 Davis-Dusenbery BN, Wu C, Hata A: Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol 2011;31:2370-2377. https://doi.org/10.1161/ATVBAHA.111.226670 |
||||
| 40 Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L: miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 2011;4:197-205. https://doi.org/10.1161/CIRCGENETICS.110.958702 |
||||
| 41 Bodine PV, Komm BS: Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord 2006;7:33-39. https://doi.org/10.1007/s11154-006-9002-4 |
||||
| 42 Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA: Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler Thromb Vasc Biol 2007;27:2589-2596. https://doi.org/10.1161/ATVBAHA.107.153668 |
||||
| 43 Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW: Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord 2006;7:1-16. https://doi.org/10.1007/s11154-006-9001-5 |
||||
| 44 Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B: The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002;108:17-29. https://doi.org/10.1016/S0092-8674(01)00622-5 |
||||
| 45 Zhu F, Friedman MS, Luo W, Woolf P, Hankenson KD: The transcription factor osterix (SP7) regulates BMP6-induced human osteoblast differentiation. J Cell Physiol 2012;227:2677-2685. https://doi.org/10.1002/jcp.23010 |
||||
| 46 Yang H, Curinga G, Giachelli CM: Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int 2004;66:2293-2299. https://doi.org/10.1111/j.1523-1755.2004.66015.x |
||||
| 47 Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM: Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 2001;89:1147-1154. https://doi.org/10.1161/hh2401.101070 |
||||
| 48 Chen NX, O'Neill KD, Duan D, Moe SM: Phosphorus and uremic serum up-regulate osteopontin expression in vascular smooth muscle cells. Kidney Int 2002;62:1724-1731. https://doi.org/10.1046/j.1523-1755.2002.00625.x |
||||
| 49 Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, Frutkin A, Dichek D, Giachelli CM: Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res 2009;104:733-741. https://doi.org/10.1161/CIRCRESAHA.108.183053 |
||||
| 50 Cozzolino M, Dusso AS, Slatopolsky E: Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. J Am Soc Nephrol 2001;12:2511-2516. https://doi.org/10.1681/ASN.V12112511 |
||||
| 51 Moe SM, O'Neill KD, Duan D, Ahmed S, Chen NX, Leapman SB, Fineberg N, Kopecky K: Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int 2002;61:638-647. https://doi.org/10.1046/j.1523-1755.2002.00170.x |
||||
| 52 Moe SM, Duan D, Doehle BP, O'Neill KD, Chen NX: Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int 2003;63:1003-1011. https://doi.org/10.1046/j.1523-1755.2003.00820.x |
||||
| 53 London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H: Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003;18:1731-1740. https://doi.org/10.1093/ndt/gfg414 |
||||
| 54 Serban KA, Rezania S, Petrusca DN, Poirier C, Cao D, Justice MJ, Patel M, Tsvetkova I, Kamocki K, Mikosz A, Schweitzer KS, Jacobson S, Cardoso A, Carlesso N, Hubbard WC, Kechris K, Dragnea B, Berdyshev EV, McClintock J, Petrache I: Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep 2016;6:31596. https://doi.org/10.1038/srep31596 |
||||
| 55 Goldsmith DJ, Covic A, Sambrook PA, Ackrill P: Vascular calcification in long-term haemodialysis patients in a single unit: a retrospective analysis. Nephron 1997;77:37-43. https://doi.org/10.1159/000190244 |
||||
| 56 Kang YH, Jin JS, Yi DW, Son SM: Bone morphogenetic protein-7 inhibits vascular calcification induced by high vitamin D in mice. Tohoku J Exp Med 2010;221:299-307. https://doi.org/10.1620/tjem.221.299 |
||||
| 57 Bas A, Lopez I, Perez J, Rodriguez M, Aguilera-Tejero E: Reversibility of calcitriol-induced medial artery calcification in rats with intact renal function. J Bone Miner Res 2006;21:484-490. https://doi.org/10.1359/JBMR.051211 |
||||
| 58 Bhat OM, Yuan X, Camus S, Salloum FN, Li PL: Abnormal Lysosomal Positioning and Small Extracellular Vesicle Secretion in Arterial Stiffening and Calcification of Mice Lacking Mucolipin 1 Gene. Int J Mol Sci 2020;21:1713. https://doi.org/10.3390/ijms21051713 |
||||
| 59 Cai Y, Wang XL, Flores AM, Lin T, Guzman RJ: Inhibition of endo-lysosomal function exacerbates vascular calcification. Sci Rep 2018;8:3377. https://doi.org/10.1038/s41598-017-17540-6 |
||||
| 60 Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, Witwer KW, Thery C: Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014;3:26913. https://doi.org/10.3402/jev.v3.26913 |
||||
| 61 Al-Nedawi K, Meehan B, Rak J: Microvesicles: messengers and mediators of tumor progression. Cell Cycle 2009;8:2014-2018. https://doi.org/10.4161/cc.8.13.8988 |
||||
| 62 Sotelo JR, Porter KR: An electron microscope study of the rat ovum. J Biophys Biochem Cytol 1959;5:327-342. https://doi.org/10.1083/jcb.5.2.327 |
||||
| 63 Akers JC, Gonda D, Kim R, Carter BS, Chen CC: Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol 2013;113:1-11. https://doi.org/10.1007/s11060-013-1084-8 |
||||
| 64 Boulanger CM, Loyer X, Rautou PE, Amabile N: Extracellular vesicles in coronary artery disease. Nat Rev Cardiol 2017;14:259-272. https://doi.org/10.1038/nrcardio.2017.7 |
||||
| 65 Tetta C, Ghigo E, Silengo L, Deregibus MC, Camussi G: Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 2013;44:11-19. https://doi.org/10.1007/s12020-012-9839-0 |
||||
| 66 van Rooij E, Olson EN: MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov 2012;11:860-872. https://doi.org/10.1038/nrd3864 |
||||
| 67 Aikawa E: Extracellular vesicles in cardiovascular disease: focus on vascular calcification. J Physiol 2016;594:2877-2880. https://doi.org/10.1113/JP272112 |
||||
| 68 Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S, Aikawa E: Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater 2016;15:335-343. https://doi.org/10.1038/nmat4519 |
||||
| 69 Oggero S, Austin-Williams S, Norling LV: The Contrasting Role of Extracellular Vesicles in Vascular Inflammation and Tissue Repair. Front Pharmacol 2019;10:1479. https://doi.org/10.3389/fphar.2019.01479 |
||||
| 70 Buendia P, Montes de Oca A, Madueno JA, Merino A, Martin-Malo A, Aljama P, Ramirez R, Rodriguez M, Carracedo J: Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J 2015;29:173-181. https://doi.org/10.1096/fj.14-249706 |
||||
| 71 Ikeda K, Souma Y, Akakabe Y, Kitamura Y, Matsuo K, Shimoda Y, Ueyama T, Matoba S, Yamada H, Okigaki M, Matsubara H: Macrophages play a unique role in the plaque calcification by enhancing the osteogenic signals exerted by vascular smooth muscle cells. Biochem Biophys Res Commun 2012;425:39-44. https://doi.org/10.1016/j.bbrc.2012.07.045 |
||||
| 72 New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E: Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res 2013;113:72-77. https://doi.org/10.1161/CIRCRESAHA.113.301036 |
||||
| 73 Alique M, Ruiz-Torres MP, Bodega G, Noci MV, Troyano N, Bohorquez L, Luna C, Luque R, Carmona A, Carracedo J, Ramirez R: Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging (Albany NY) 2017;9:778-789. https://doi.org/10.18632/aging.101191 |
||||
| 74 Jansen F, Xiang X, Werner N: Role and function of extracellular vesicles in calcific aortic valve disease. Eur Heart J 2017;38:2714-2716. https://doi.org/10.1093/eurheartj/ehx477 |
||||
| 75 Deng L, Blanco FJ, Stevens H, Lu R, Caudrillier A, McBride M, McClure JD, Grant J, Thomas M, Frid M, Stenmark K, White K, Seto AG, Morrell NW, Bradshaw AC, MacLean MR, Baker AH: MicroRNA-143 Activation Regulates Smooth Muscle and Endothelial Cell Crosstalk in Pulmonary Arterial Hypertension. Circ Res 2015;117:870-883. https://doi.org/10.1161/CIRCRESAHA.115.306806 |
||||
| 76 Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M: Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008;319:1244-1247. https://doi.org/10.1126/science.1153124 |
||||
| 77 Reynolds JL, Skepper JN, McNair R, Kasama T, Gupta K, Weissberg PL, Jahnen-Dechent W, Shanahan CM: Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol 2005;16:2920-2930. https://doi.org/10.1681/ASN.2004100895 |
||||
| 78 Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM: Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res 2011;109:e1-12. https://doi.org/10.1161/CIRCRESAHA.110.238808 |
||||
| 79 Shroff RC, Shah V, Hiorns MP, Schoppet M, Hofbauer LC, Hawa G, Schurgers LJ, Singhal A, Merryweather I, Brogan P, Shanahan C, Deanfield J, Rees L: The circulating calcification inhibitors, fetuin-A and osteoprotegerin, but not matrix Gla protein, are associated with vascular stiffness and calcification in children on dialysis. Nephrol Dial Transplant 2008;23:3263-3271. https://doi.org/10.1093/ndt/gfn226 |
||||
| 80 Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, Kjolby M, Rogers M, Michel T, Shibasaki M, Hagita S, Kramann R, Rader DJ, Libby P, Singh SA, Aikawa E: Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest 2016;126:1323-1336. https://doi.org/10.1172/JCI80851 |
||||
| 81 Chae YM, Heo SH, Kim JY, Lee JM, Ryoo HM, Cho JY: Upregulation of smpd3 via BMP2 stimulation and Runx2. BMB Rep 2009;42:86-90. https://doi.org/10.5483/BMBRep.2009.42.2.086 |
||||
| 82 Kakoi H, Maeda S, Shinohara N, Matsuyama K, Imamura K, Kawamura I, Nagano S, Setoguchi T, Yokouchi M, Ishidou Y, Komiya S: Bone morphogenic protein (BMP) signaling up-regulates neutral sphingomyelinase 2 to suppress chondrocyte maturation via the Akt protein signaling pathway as a negative feedback mechanism. J Biol Chem 2014;289:8135-8150. https://doi.org/10.1074/jbc.M113.509331 |
||||
| 83 Leroyer AS, Isobe H, Leseche G, Castier Y, Wassef M, Mallat Z, Binder BR, Tedgui A, Boulanger CM: Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol 2007;49:772-777. https://doi.org/10.1016/j.jacc.2006.10.053 |
||||
| 84 Kapustin AN, Schoppet M, Schurgers LJ, Reynolds JL, McNair R, Heiss A, Jahnen-Dechent W, Hackeng TM, Schlieper G, Harrison P, Shanahan CM: Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arterioscler Thromb Vasc Biol 2017;37:e22-e32. https://doi.org/10.1161/ATVBAHA.116.308886 |
||||
| 85 Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD: Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem 2013;288:10093-10099. https://doi.org/10.1074/jbc.C112.444562 |
||||
| 86 Li C, Cui Y, Luan J, Zhou X, Li H, Wang H, Shi L, Han J: Tenascin C affects mineralization of SaOS2 osteoblast-like cells through matrix vesicles. Drug Discov Ther 2016;10:82-87. https://doi.org/10.5582/ddt.2016.01009 |
||||
| 87 de Jong OG, van Balkom BW, Gremmels H, Verhaar MC: Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2. J Cell Mol Med 2016;20:342-350. https://doi.org/10.1111/jcmm.12730 |
||||
| 88 Mu W, Rana S, Zoller M: Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia (New York, NY) 2013;15:875-887. https://doi.org/10.1593/neo.13786 |
||||
| 89 Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM: Directional cell movement through tissues is controlled by exosome secretion. Nat Commun 2015;6:7164. https://doi.org/10.1038/ncomms8164 |
||||
| 90 Dolo V, D'Ascenzo S, Violini S, Pompucci L, Festuccia C, Ginestra A, Vittorelli ML, Canevari S, Pavan A: Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clin Exp Metastasis 1999;17:131-140. https://doi.org/10.1023/A:1006500406240 |
||||
| 91 D'Souza-Schorey C, Clancy JW: Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev 2012;26:1287-1299. https://doi.org/10.1101/gad.192351.112 |
||||
| 92 Gerlach JQ, Griffin MD: Getting to know the extracellular vesicle glycome. Mol Biosyst 2016;12:1071-1081. https://doi.org/10.1039/C5MB00835B |
||||
| 93 Schmidt JR, Kliemt S, Preissler C, Moeller S, von Bergen M, Hempel U, Kalkhof S: Osteoblast-released Matrix Vesicles, Regulation of Activity and Composition by Sulfated and Non-sulfated Glycosaminoglycans. Mol Cell Proteomics 2016;15:558-572. https://doi.org/10.1074/mcp.M115.049718 |
||||
| 94 Wu LN, Genge BR, Wuthier RE: Association between proteoglycans and matrix vesicles in the extracellular matrix of growth plate cartilage. J Biol Chem 1991;266:1187-1194. https://doi.org/10.1016/S0021-9258(17)35300-0 |
||||
| 95 Rilla K, Pasonen-Seppanen S, Deen AJ, Koistinen VVT, Wojciechowski S, Oikari S, Karna R, Bart G, Torronen K, Tammi RH, Tammi MI: Hyaluronan production enhances shedding of plasma membrane-derived microvesicles. Exp Cell Res 2013;319:2006-2018. https://doi.org/10.1016/j.yexcr.2013.05.021 |
||||
| 96 Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M: Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A 2013;110:17380-17385. https://doi.org/10.1073/pnas.1304266110 |
||||
| 97 Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD: Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem 2013;288:10093-10099. https://doi.org/10.1074/jbc.C112.444562 |
||||
| 98 Genge BR, Wu LN, Wuthier RE: Identification of phospholipid-dependent calcium-binding proteins as constituents of matrix vesicles. J Biol Chem 1989;264:10917-10921. https://doi.org/10.1016/S0021-9258(18)81708-2 |
||||
| 99 New SE, Aikawa E: Role of extracellular vesicles in de novo mineralization: an additional novel mechanism of cardiovascular calcification. Arterioscler Thromb Vasc Biol 2013;33:1753-1758. https://doi.org/10.1161/ATVBAHA.112.300128 |
||||
| 100 Kirsch T, Pfaffle M: Selective binding of anchorin CII (annexin V) to type II and X collagen and to chondrocalcin (C-propeptide of type II collagen). Implications for anchoring function between matrix vesicles and matrix proteins. FEBS Lett 1992;310:143-147. https://doi.org/10.1016/0014-5793(92)81316-E |
||||
| 101 Appelqvist H, Waster P, Kagedal K, Ollinger K: The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013;5:214-226. https://doi.org/10.1093/jmcb/mjt022 |
||||
| 102 Appelmans F, Wattiaux R, De Duve C: Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochem J 1955;59:438-445. https://doi.org/10.1042/bj0590438 |
||||
| 103 Ferreira CR, Gahl WA: Lysosomal storage diseases. Transl Sci Rare Dis 2017;2:1-71. https://doi.org/10.3233/TRD-160005 |
||||
| 104 Braunlin E, Orchard PJ, Whitley CB, Schroeder L, Reed RC, Manivel JC: Unexpected coronary artery findings in mucopolysaccharidosis. Report of four cases and literature review. Cardiovasc Pathol 2014;23:145-151. https://doi.org/10.1016/j.carpath.2014.01.001 |
||||
| 105 Wendelhag I, Gustavsson T, Suurkula M, Berglund G, Wikstrand J: Ultrasound measurement of wall thickness in the carotid artery: fundamental principles and description of a computerized analysing system. Clin Physiol 1991;11:565-577. https://doi.org/10.1111/j.1475-097X.1991.tb00676.x |
||||
| 106 Wang RY, Covault KK, Halcrow EM, Gardner AJ, Cao X, Newcomb RL, Dauben RD, Chang AC: Carotid intima-media thickness is increased in patients with mucopolysaccharidoses. Mol Genet Metab 2011;104:592-596. https://doi.org/10.1016/j.ymgme.2011.09.004 |
||||
| 107 Wang RY, Braunlin EA, Rudser KD, Dengel DR, Metzig AM, Covault KK, Polgreen LE, Shapiro E, Steinberger J, Kelly AS: Carotid intima-media thickness is increased in patients with treated mucopolysaccharidosis types I and II, and correlates with arterial stiffness. Mol Genet Metab 2014;111:128-132. https://doi.org/10.1016/j.ymgme.2013.11.001 |
||||
| 108 Metcalf JA, Linders B, Wu S, Bigg P, O'Donnell P, Sleeper MM, Whyte MP, Haskins M, Ponder KP: Upregulation of elastase activity in aorta in mucopolysaccharidosis I and VII dogs may be due to increased cytokine expression. Mol Genet Metab 2010;99:396-407. https://doi.org/10.1016/j.ymgme.2009.12.003 |
||||
| 109 Lyons JA, Dickson PI, Wall JS, Passage MB, Ellinwood NM, Kakkis ED, McEntee MF: Arterial pathology in canine mucopolysaccharidosis-I and response to therapy. Lab Invest 2011;91:665-674. https://doi.org/10.1038/labinvest.2011.7 |
||||
| 110 Khalid O, Vera MU, Gordts PL, Ellinwood NM, Schwartz PH, Dickson PI, Esko JD, Wang RY: Immune-Mediated Inflammation May Contribute to the Pathogenesis of Cardiovascular Disease in Mucopolysaccharidosis Type I. PLoS One 2016;11:e0150850. https://doi.org/10.1371/journal.pone.0150850 |
||||
| 111 Simonaro CM, Tomatsu S, Sikora T, Kubaski F, Frohbergh M, Guevara JM, Wang RY, Vera M, Kang JL, Smith LJ, Schuchman EH, Haskins ME: Pentosan Polysulfate: Oral Versus Subcutaneous Injection in Mucopolysaccharidosis Type I Dogs. PLoS One 2016;11:e0153136. https://doi.org/10.1371/journal.pone.0153136 |
||||
| 112 Syeda B, Gottsauner-Wolf M, Denk S, Pichler P, Khorsand A, Glogar D: Arterial compliance: a diagnostic marker for atherosclerotic plaque burden? Am J Hypertens 2003;16:356-362. https://doi.org/10.1016/S0895-7061(03)00037-2 |
||||
| 113 Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L: Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med 1967;276:1163-1167. https://doi.org/10.1056/NEJM196705252762101 |
||||
| 114 Kint JA: Fabry's disease: alpha-galactosidase deficiency. Science 1970;167:1268-1269. https://doi.org/10.1126/science.167.3922.1268 |
||||
| 115 Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE: Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab 2010;99:99-108. https://doi.org/10.1016/j.ymgme.2009.10.004 |
||||
| 116 McCarthy CG, Wenceslau CF, Calmasini FB, Klee NS, Brands MW, Joe B, Webb RC: Reconstitution of autophagy ameliorates vascular function and arterial stiffening in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2019;317:H1013-H1027. https://doi.org/10.1152/ajpheart.00227.2019 |
||||
| 117 Lane HA, Smith JC, Davies JS: Noninvasive assessment of preclinical atherosclerosis. Vasc Health Risk Manag 2006;2:19-30. https://doi.org/10.2147/vhrm.2006.2.1.19 |
||||
| 118 Mallika V, Goswami B, Rajappa M: Atherosclerosis pathophysiology and the role of novel risk factors: a clinicobiochemical perspective. Angiology 2007;58:513-522. https://doi.org/10.1177/0003319707303443 |
||||
| 119 Goldstein JL, Brown MS: Atherosclerosis: the low-density lipoprotein receptor hypothesis. Metabolism 1977;26:1257-1275. https://doi.org/10.1016/0026-0495(77)90119-6 |
||||
| 120 Ross R: The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993;362:801-809. https://doi.org/10.1038/362801a0 |
||||
| 121 Libby P, Ridker PM: Novel inflammatory markers of coronary risk: theory versus practice. Circulation 1999;100:1148-1150. https://doi.org/10.1161/01.CIR.100.11.1148 |
||||
| 122 Kutuk O, Basaga H: Inflammation meets oxidation: NF-kappaB as a mediator of initial lesion development in atherosclerosis. Trends Mol Med 2003;9:549-557. https://doi.org/10.1016/j.molmed.2003.10.007 |
||||
| 123 Mantovani A, Garlanda C, Locati M: Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 2009;29:1419-1423. https://doi.org/10.1161/ATVBAHA.108.180497 |
||||
| 124 Barlic J, Zhang Y, Murphy PM: Atherogenic lipids induce adhesion of human coronary artery smooth muscle cells to macrophages by up-regulating chemokine CX3CL1 on smooth muscle cells in a TNFalpha-NFkappaB-dependent manner. J Biol Chem 2007;282:19167-19176. https://doi.org/10.1074/jbc.M701642200 |
||||
| 125 Packard RR, Libby P: Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem 2008;54:24-38. https://doi.org/10.1373/clinchem.2007.097360 |
||||
| 126 Raffetto JD, Khalil RA: Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 2008;75:346-359. https://doi.org/10.1016/j.bcp.2007.07.004 |
||||
| 127 Libby P: Atherosclerosis: disease biology affecting the coronary vasculature. Am J Cardiol 2006;98:3Q-9Q. https://doi.org/10.1016/j.amjcard.2006.09.020 |
||||
| 128 Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA: Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 2007;117:1782-1793. https://doi.org/10.1172/JCI27523 |
||||
| 129 Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V: Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004;119:753-766. https://doi.org/10.1016/j.cell.2004.11.038 |
||||
| 130 Xu K, Yang Y, Yan M, Zhan J, Fu X, Zheng X: Autophagy plays a protective role in free cholesterol overload-induced death of smooth muscle cells. J Lipid Res 2010;51:2581-2590. https://doi.org/10.1194/jlr.M005702 |
||||
| 131 Jia G, Cheng G, Gangahar DM, Agrawal DK: Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 2006;84:448-454. https://doi.org/10.1111/j.1440-1711.2006.01454.x |
||||
| 132 Martinon F, Mayor A, Tschopp J: The inflammasomes: guardians of the body. Annu Rev Immunol 2009;27:229-265. https://doi.org/10.1146/annurev.immunol.021908.132715 |
||||
| 133 Jia G, Cheng G, Agrawal DK: Autophagy of vascular smooth muscle cells in atherosclerotic lesions. Autophagy 2007;3:63-64. https://doi.org/10.4161/auto.3427 |
||||
| 134 Schrijvers DM, De Meyer GR, Herman AG, Martinet W: Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res 2007;73:470-480. https://doi.org/10.1016/j.cardiores.2006.09.005 |
||||
| 135 Zhang Y, Xu M, Xia M, Li X, Boini KM, Wang M, Gulbins E, Ratz PH, Li PL: Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res 2014;102:68-78. https://doi.org/10.1093/cvr/cvu011 |
||||
| 136 Bao JX, Zhang QF, Wang M, Xia M, Boini KM, Gulbins E, Zhang Y, Li PL: Implication of CD38 gene in autophagic degradation of collagen I in mouse coronary arterial myocytes. Front Biosci (Landmark Ed) 2017;22:558-569. https://doi.org/10.2741/4502 |
||||
| 137 Li X, Xu M, Pitzer AL, Xia M, Boini KM, Li PL, Zhang Y: Control of autophagy maturation by acid sphingomyelinase in mouse coronary arterial smooth muscle cells: protective role in atherosclerosis. J Mol Med (Berl) 2014;92:473-485. https://doi.org/10.1007/s00109-014-1120-y |
||||
| 138 Yuan X, Bhat OM, Lohner H, Li N, Zhang Y, Li PL: Inhibitory effects of growth differentiation factor 11 on autophagy deficiency-induced dedifferentiation of arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 2019;316:H345-H356. https://doi.org/10.1152/ajpheart.00342.2018 |
||||
| 139 Ikeda S, Ushio-Fukai M, Zuo L, Tojo T, Dikalov S, Patrushev NA, Alexander RW: Novel role of ARF6 in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 2005;96:467-475. https://doi.org/10.1161/01.RES.0000158286.51045.16 |
||||
| 140 Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C: Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003;18:655-664. https://doi.org/10.1016/S1074-7613(03)00092-X |
||||
| 141 Jin S, Yi F, Li PL: Contribution of lysosomal vesicles to the formation of lipid raft redox signaling platforms in endothelial cells. Antioxidants & redox signaling 2007;9:1417-1426. https://doi.org/10.1089/ars.2007.1660 |
||||
| 142 Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW: Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arterioscler Thromb Vasc Biol 2005;25:1824-1830. https://doi.org/10.1161/01.ATV.0000175295.09607.18 |
||||
| 143 Yang B, Rizzo V: TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 2007;292:H954-962. https://doi.org/10.1152/ajpheart.00758.2006 |
||||
| 144 Cai H: NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res 2005;96:818-822. https://doi.org/10.1161/01.RES.0000163631.07205.fb |
||||
| 145 Suzuki YJ, Ford GD: Redox regulation of signal transduction in cardiac and smooth muscle. J Mol Cell Cardiol 1999;31:345-353. https://doi.org/10.1006/jmcc.1998.0872 |
||||
| 146 Eum SY, Andras I, Hennig B, Toborek M: NADPH oxidase and lipid raft-associated redox signaling are required for PCB153-induced upregulation of cell adhesion molecules in human brain endothelial cells. Toxicol Appl Pharmacol 2009;240:299-305. https://doi.org/10.1016/j.taap.2009.07.022 |
||||
| 147 Peshavariya H, Dusting GJ, Di Bartolo B, Rye KA, Barter PJ, Jiang F: Reconstituted high-density lipoprotein suppresses leukocyte NADPH oxidase activation by disrupting lipid rafts. Free Radic Res 2009;43:772-782. https://doi.org/10.1080/10715760903045304 |
||||
| 148 Jin S, Yi F, Zhang F, Poklis JL, Li PL: Lysosomal targeting and trafficking of acid sphingomyelinase to lipid raft platforms in coronary endothelial cells. Arterioscler Thromb Vasc Biol 2008;28:2056-2062. https://doi.org/10.1161/ATVBAHA.108.172478 |
||||
| 149 Jin S, Zhang Y, Yi F, Li PL: Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol 2008;28:485-490. https://doi.org/10.1161/ATVBAHA.107.159772 |
||||
| 150 Huynh C, Roth D, Ward DM, Kaplan J, Andrews NW: Defective lysosomal exocytosis and plasma membrane repair in Chediak-Higashi/beige cells. Proc Natl Acad Sci U S A 2004;101:16795-16800. https://doi.org/10.1073/pnas.0405905101 |
||||
| 151 Jaiswal JK, Andrews NW, Simon SM: Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol 2002;159:625-635. https://doi.org/10.1083/jcb.200208154 |
||||
| 152 Xia M, Zhang C, Boini KM, Thacker AM, Li PL: Membrane raft-lysosome redox signalling platforms in coronary endothelial dysfunction induced by adipokine visfatin. Cardiovasc Res 2011;89:401-409. https://doi.org/10.1093/cvr/cvq286 |
||||
| 153 Qiu H, Edmunds T, Baker-Malcolm J, Karey KP, Estes S, Schwarz C, Hughes H, Van Patten SM: Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J Biol Chem 2003;278:32744-32752. https://doi.org/10.1074/jbc.M303022200 |
||||
| 154 Zhang AY, Yi F, Jin S, Xia M, Chen QZ, Gulbins E, Li PL: Acid sphingomyelinase and its redox amplification in formation of lipid raft redox signaling platforms in endothelial cells. Antioxid Redox Signal 2007;9:817-828. https://doi.org/10.1089/ars.2007.1509 |
||||
| 155 Bao JX, Xia M, Poklis JL, Han WQ, Brimson C, Li PL: Triggering role of acid sphingomyelinase in endothelial lysosome-membrane fusion and dysfunction in coronary arteries. Am J Physiol Heart Circ Physiol 2010;298:H992-H1002. https://doi.org/10.1152/ajpheart.00958.2009 |
||||
| 156 Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, Li PL: Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol 2017;13:336-344. https://doi.org/10.1016/j.redox.2017.06.004 |
||||
| 157 Yuan X, Wang L, Bhat OM, Lohner H, Li PL: Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: Antioxidant action of butyrate. Redox Biol 2018;16:21-31. https://doi.org/10.1016/j.redox.2018.02.007 |
||||
| 158 Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM: Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004;430:213-218. https://doi.org/10.1038/nature02664 |
||||
| 159 Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J: Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010;11:136-140. https://doi.org/10.1038/ni.1831 |
||||
| 160 Dinarello CA, Donath MY, Mandrup-Poulsen T: Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010;17:314-321. https://doi.org/10.1097/MED.0b013e32833bf6dc |
||||
| 161 Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, Derfalvi B, Benjaponpitak S, Vesely R, Sauvain MJ, Oertle S, Allen R, Morgan G, Borkhardt A, Hill C, Gardner-Medwin J, Fischer A, de Saint Basile G: Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood 2004;103:2809-2815. https://doi.org/10.1182/blood-2003-07-2531 |
||||
| 162 Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E: NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357-1361. https://doi.org/10.1038/nature08938 |
||||
| 163 Abais JM, Xia M, Zhang Y, Boini KM, Li PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal 2015;22:1111-1129. https://doi.org/10.1089/ars.2014.5994 |
||||
| 164 Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006;440:237-241. https://doi.org/10.1038/nature04516 |
||||
| 165 Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT: NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674-678. https://doi.org/10.1038/nature11729 |
||||
| 166 Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL: Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One 2014;9:e87552. https://doi.org/10.1371/journal.pone.0087552 |
||||
| 167 Zhang C, Boini KM, Xia M, Abais JM, Li X, Liu Q, Li PL: Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012;60:154-162. https://doi.org/10.1161/HYPERTENSIONAHA.111.189688 |
||||
| 168 Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL: Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol 2014;184:1617-1628. https://doi.org/10.1016/j.ajpath.2014.01.032 |
||||
| 169 Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, Li PL: Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta 2015;1853:396-408. https://doi.org/10.1016/j.bbamcr.2014.11.012 |
||||
| 170 Zhang Y, Li X, Pitzer AL, Chen Y, Wang L, Li PL: Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: beyond inflammation. Antioxid Redox Signal 2015;22:1084-1096. https://doi.org/10.1089/ars.2014.5978 |
||||
| 171 Yuan X, Bhat OM, Meng N, Lohner H, Li PL: Protective Role of Autophagy in Nlrp3 Inflammasome Activation and Medial Thickening of Mouse Coronary Arteries. Am J Pathol 2018;188:2948-2959. https://doi.org/10.1016/j.ajpath.2018.08.014 |
||||
| 172 Yuan X, Bhat OM, Lohner H, Zhang Y, Li PL: Endothelial acid ceramidase in exosome-mediated release of NLRP3 inflammasome products during hyperglycemia: Evidence from endothelium-specific deletion of Asah1 gene. Biochim Biophys Acta Mol Cell Biol Lipids 2019;1864:158532. https://doi.org/10.1016/j.bbalip.2019.158532 |
||||
| 173 Anderson RG, Vasile E, Mello RJ, Brown MS, Goldstein JL: Immunocytochemical visualization of coated pits and vesicles in human fibroblasts: relation to low density lipoprotein receptor distribution. Cell 1978;15:919-933. https://doi.org/10.1016/0092-8674(78)90276-3 |
||||
| 174 Daniels TF, Killinger KM, Michal JJ, Wright RW, Jr., Jiang Z: Lipoproteins, cholesterol homeostasis and cardiac health. Int J Biol Sci 2009;5:474-488. https://doi.org/10.7150/ijbs.5.474 |
||||
| 175 Xu X, Yuan X, Li N, Dewey WL, Li PL, Zhang F: Lysosomal cholesterol accumulation in macrophages leading to coronary atherosclerosis in CD38(-/-) mice. J Cell Mol Med 2016;20:1001-1013. https://doi.org/10.1111/jcmm.12788 |
||||
| 176 Jerome WG, Yancey PG: The role of microscopy in understanding atherosclerotic lysosomal lipid metabolism. Microsc Microanal 2003;9:54-67. https://doi.org/10.1017/S1431927603030010 |
||||
| 177 Miller BF, Kothari HV: Increased activity of lysosomal enzymes in human atherosclerotic aortas. Exp Mol Pathol 1969;10:288-294. https://doi.org/10.1016/0014-4800(69)90058-6 |
||||
| 178 Fowler S, Berberian PA, Shio H, Goldfischer S, Wolinsky H: Characterization of cell populations isolated from aortas of rhesus monkeys with experimental atherosclerosis. Circ Res 1980;46:520-530. https://doi.org/10.1161/01.RES.46.4.520 |
||||
| 179 Bach G: Mucolipidosis type IV. Mol Genet Metab 2001;73:197-203. https://doi.org/10.1006/mgme.2001.3195 |
||||
| 180 Slaugenhaupt SA: The molecular basis of mucolipidosis type IV. Curr Mol Med 2002;2:445-450. https://doi.org/10.2174/1566524023362276 |
||||
| 181 Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ: Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 1997;277:232-235. https://doi.org/10.1126/science.277.5323.232 |
||||