×
![]()
Corresponding Author: Amit Mishra
Cellular and Molecular Neurobiology Unit, Indian Institute of Technology Jodhpur, Jodhpur, Rajasthan 342037 (India)
Tel. +91-291-2801206 , E-Mail amit@iitj.ac.in
Proteasome Based Molecular Strategies Against Improper Cellular Proliferation
Ankur Rakesh Dubeya Sumit Kingera Yuvraj Anandrao Jagtapa Som Mohanlal Patwaa Prashant Kumara Sarika Singhb Amit Kumarc Awanish Kumard Amit Mishraa
aCellular and Molecular Neurobiology Unit, Indian Institute of Technology Jodhpur, Jodhpur, Rajasthan, India, bDepartment of Neuroscience and Ageing Biology and Division of Toxicology and Experimental Medicine, CSIR-Central Drug Research Institute, Lucknow, Uttar Pradesh, India, cDiscipline of Biosciences and Biomedical Engineering, Indian Institute of Technology Indore, Indore, Madhya Pradesh, India, dDepartment of Biotechnology, National Institute of Technology, Raipur, Chhattisgarh, India
Introduction
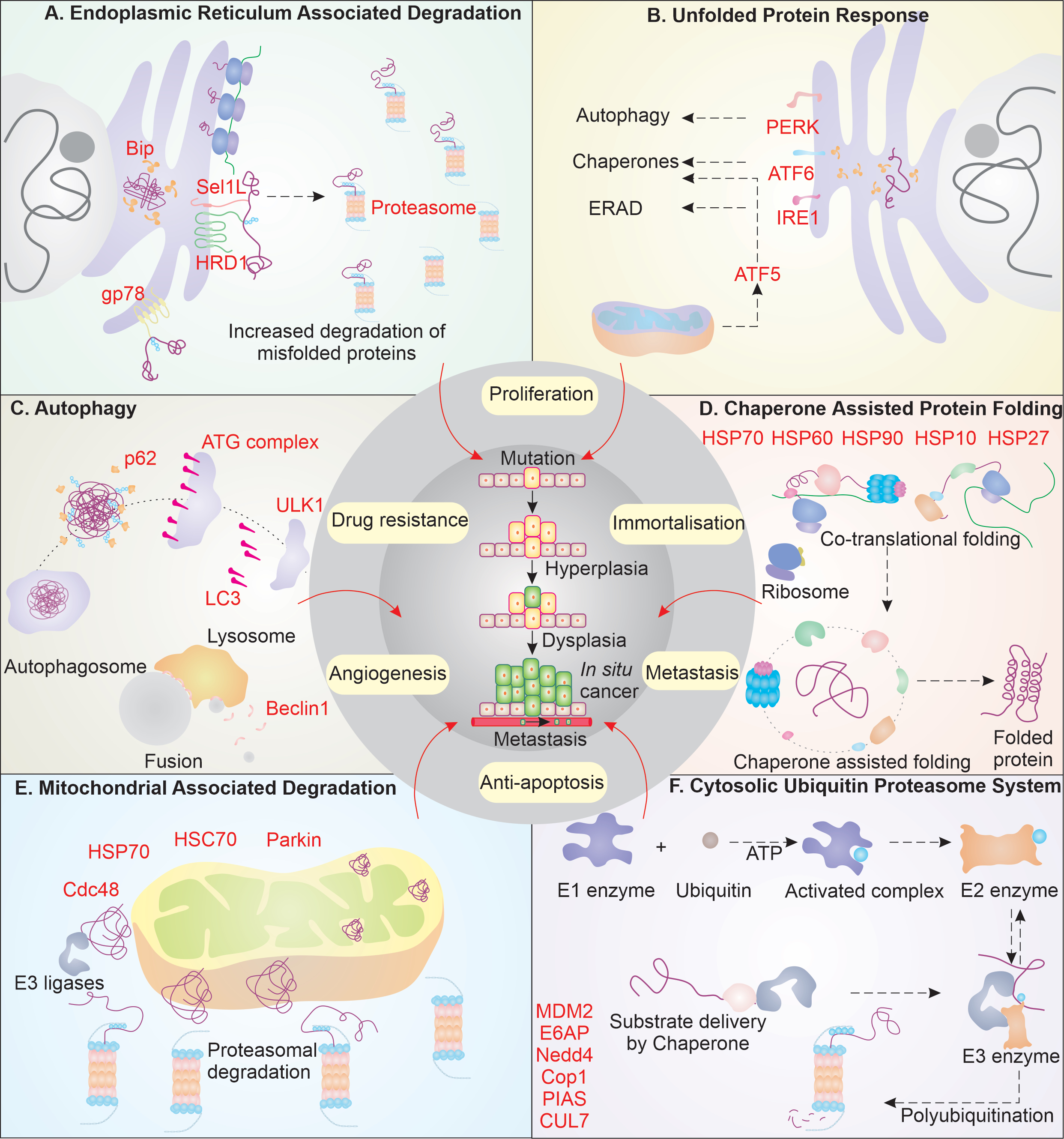
Cancer, being a very complex form of disorder, displays true nature of outcome emerging from working over the balance in cellular system. The history of cancer study has been a long journey from the first known recorded case in papyrus of Edwin Smith in 3000 B.C. to current understanding of cancer [1]. Several hypothesis and theories laid by different scientists like Hippocrates, Paracelsus, and Boerhaave have narrowed down the cause of disease from black bile to viruses, improving our earliest understanding [1]. Today, we have been able to outline the cancer cell characteristics and lay down its major hallmarks [2, 3]. David Hanseman indicated that the cancer cells have altered chromatin content and could undergo anaplasia to have rapid division characteristics [4]. The damaged chromatin in normal cells could result in extensive cellular damage; however, cancer cells are seen to survive despite such extensive DNA damage. How do they manage the extended damages and still persist the high proliferation rate? Often the damage in DNA manifests truncated, mutant or misfolded proteins, which are compromised in performing their physiological functions, and may lead to the phenomenon of protein aggregation. To avoid or deal with such conditions, cancer cells are found to have upregulated elements of system governing the overall quality of protein (Fig. 1). In this review, we have tried to lay down the mechanistic pathways of the cancer cell protein quality control, and propose how their diverse functions aid in cancer cells survivorship.
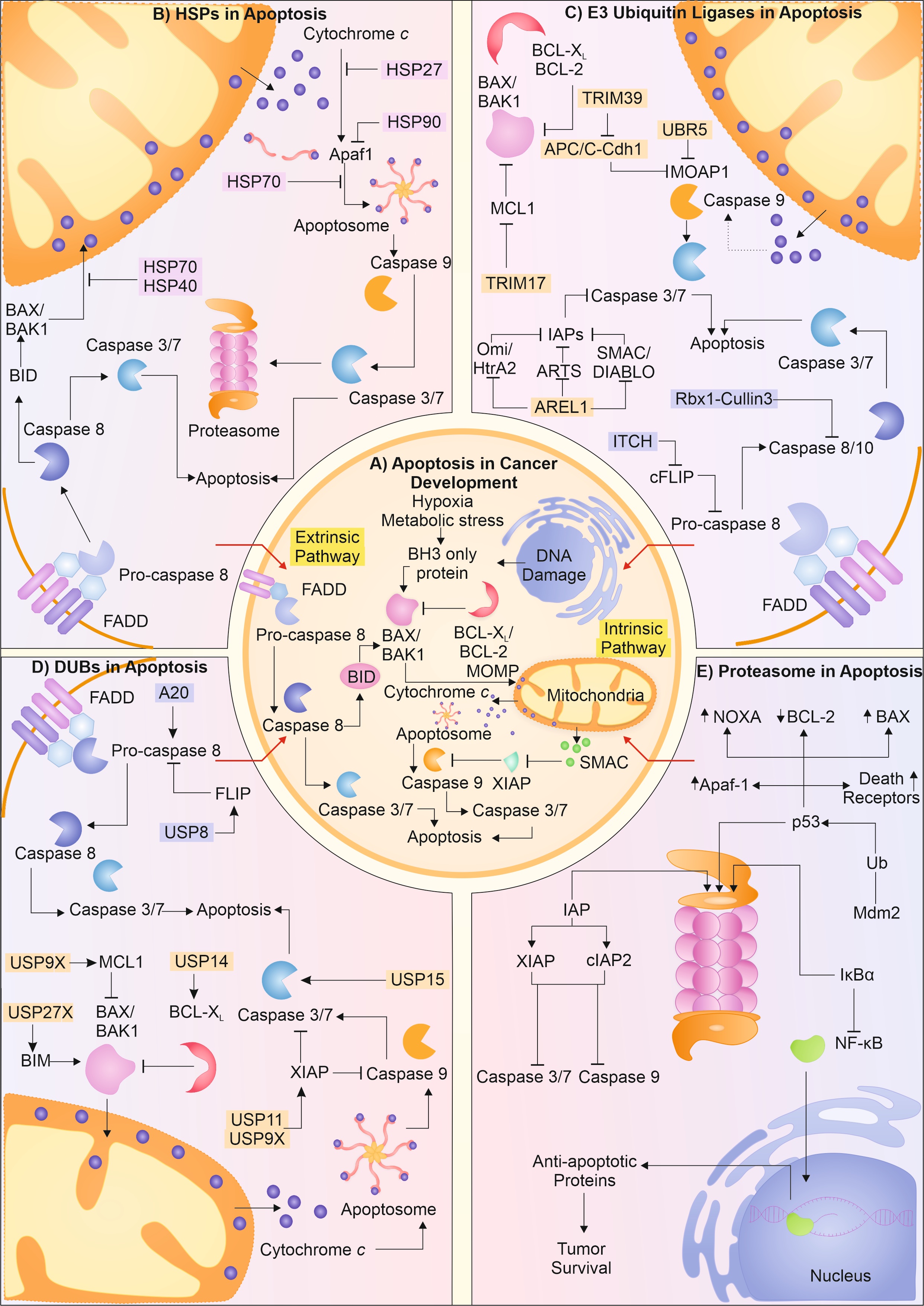
The synthesis of protein and its subsequent folding into native conformation is an outcome of vigilant action of heat shock proteins (HSPs)/chaperones. Chaperones and chaperonins principally identify and help modulate the interaction between the hydrophobic residues in polypeptide chains [5]. Largely, the function of chaperones complements the physiological functions of several key elements involved in pathways that help in cancer progression. For instance, HSP70, one of the most versatile family of chaperones, is greatly enhanced in cancer cells. It is known to perform critical functions in mediating the hypoxia response, metastasis, and extracellular immunogenic role [6]. HSP70 and other co-chaperones also help HSP90 identify the substrates and mediate their function, avoiding their aggregation. HSP90 is also seen to be highly upregulated in cancer cells, helping in cancer progression, and avoiding activating apoptosis [7]. Besides, HSPs60 and 10 are reported to mediate the folding of mitochondrial proteins; HSP10 is also reported to stall the activation of DNA damage-induced apoptosis and hence, aid in tumor progression [8-10]. Along with the classical HSPs, HSP27 is also found to be in high amounts in cancer cells. These form complexes by self-associating and try to modulate the hydrophobic interactions. HSP27 is reported to enhance tumor progression, metastasis, and help in mediating drug resistance [11, 12].
Endoplasmic reticulum (ER) performs vital function in maintaining the quality of protein, and in the case of protein misfolding, the quality control at ER mediates the misfolded protein response. ER houses two major responses to misfolding of protein viz. the unfolded protein response (UPR) and ER-associated degradation (ERAD) [13]. The proteins with defect in folding are first recognized by glucose regulated protein 78 (GRP78; Bip). It is known to control activation of the major ER membrane activators of UPR i.e., PERK, ATF6, and IRE1. Due to an increase in number of misfolded proteins, Bip is released from these activators of UPR, initiating this pathway. In case of cancer cells, both the Bip and the activators of UPR are found to be upregulated [14]. Activation of PERK leads to initiation of autophagy, whereas activation of ATF6 upregulates the chaperone expression to aid the refolding of proteins [15, 16]. Besides ER, mitochondria are also known to sense the build-up of misfolded proteins. Therefore, mitochondria mediate the mitochondrial UPR (mtUPR) and increase the expression of chaperones by activating ATF5 [17]. In case the attempts to refold the protein molecules fail, IRE1 is activated at ER, which helps in activating the ERAD pathway of protein degradation [18]. These key molecules are reported to be enhanced in cancer cells, which, apart from regulating the protein quality, help in cell survival, metastasis, tumor progression, and resistance to drugs.
The UPR-mediated activation of ERAD pathway is another key mechanism in managing the misfolded protein accumulation at ER. After the multiple attempts of refolding by chaperones and other folding proteins at ER, if the misfolded protein fails to fold, it is targeted for degradation by ERAD. The misfolded protein is transported from the ER into the cytosol at its cytosolic phase, where they are directed to proteasomal-linked degradation by E3 ligases. In cancer cells, HRD1, which is one of the main ER-associated E3 ligases in ERAD, is found to be highly upregulated. HRD1, with the aid of Sel1L (helps in translocation of protein to cytosol from lumen of ER), mediates polyubiquitination of misfolded protein clients [19, 20].
Glycoprotein 78 (gp78; AMFR, Autocrine motility factor receptor) is also known to be greatly expressed in cancer pathologies [21]. Similar to HRD1-Sel1L, gp78 also degrades misfolded protein and helps in cancer progression and metastasis [22]. The demolition of misfolded proteins and regulatory functions of ERAD is finally completed with the function of proteasome, which is enhanced in multiple cancer types [23].
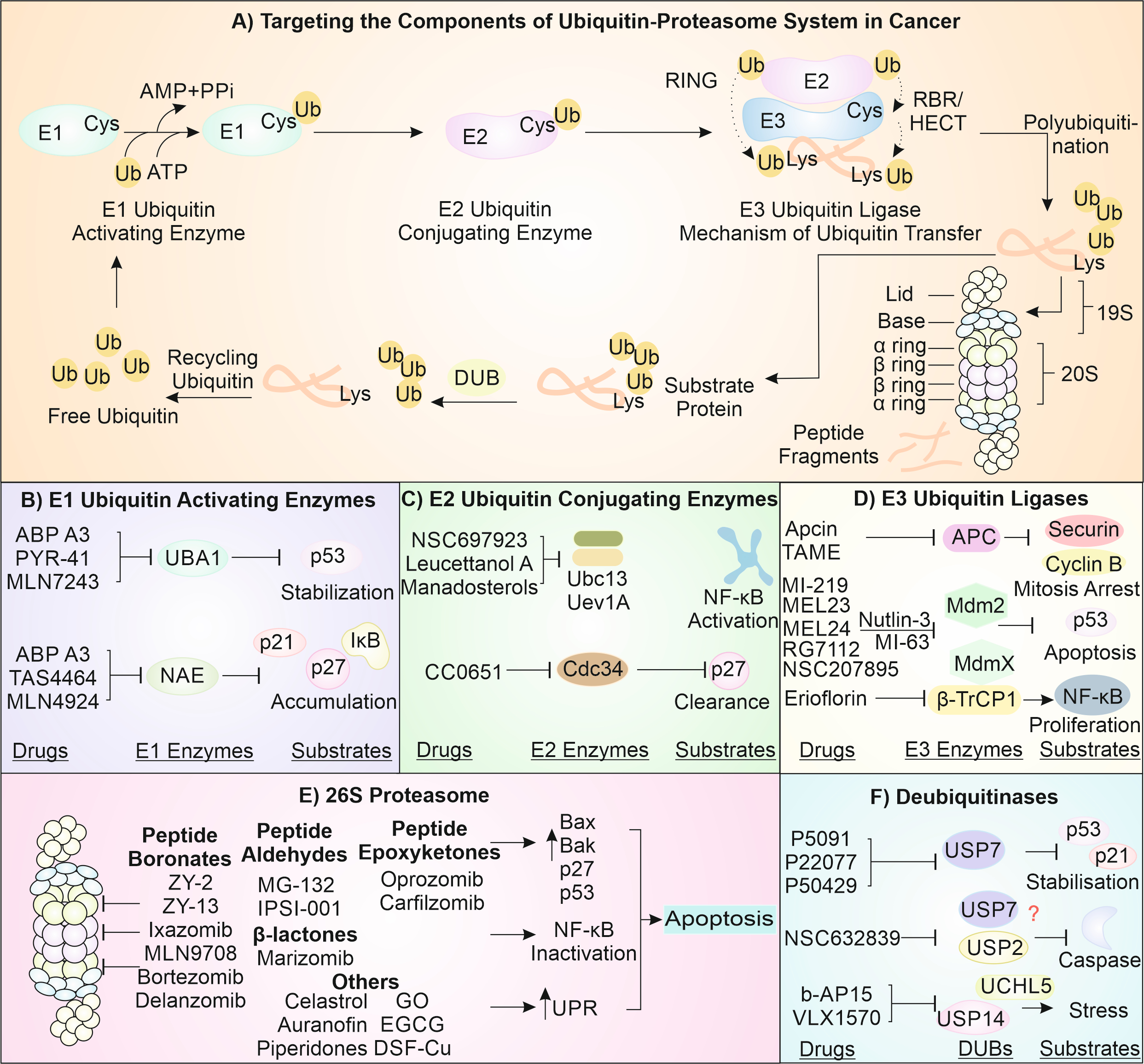
The UPS mainly manages the overall health of the cytosolic proteins and governs different cytosolic mechanisms. The basic mechanism of the system includes the addition of an activated ubiquitin chain onto a client protein. This is performed by coordinated activity of key enzymes viz. E1, E2, and E3 [24]. The E1 ubiquitin-activating enzyme helps to activate the ubiquitin by mediating its C-terminus acyl adenylation with ATP [25]. It is then shifted to E2 conjugating enzyme, which can then further interact with the E3 ligase [26]. E3 ligases are the most versatile member of the squad and have the ability to specifically identify the substrate as well as transfer the ubiquitin upon substrate [27]. The identification of substrate by E3 ligases is often facilitated by chaperones which, in a way, target the misfolded client protein for degradation by transferring them to E3s [28].
In cancer cells, several E3 enzymes are reported to be highly upregulated, and they help in throwing off the cytological balance to establish immortality and cancerous nature. For instance, the molecular aspects of cancer begin with the supression of p53 (the guardian of the genome), which is mediated by MDM2, Cop1, and Cul7-based E3 ligases. MDM2, Cop1, and Cul7 are overexpressed in cells of tumor and help in their progression and drug resistivity [29-32]. Similarly, tumor cell metastasis is inhibited by NDRG1, a growth promoting factor, which suppresses metastasis, and E6AP E3 ligase is reported to promote metastasis by degrading NDRG1 [33]. Nedd4 and similar proteins, normally involved in vesicle trafficking and protein translocation, help the cancer progression by modulating several signaling pathways driven by growth factors [34]. The ERK-mediated growth promotion is also modulated by the action of PIAS1 E3 ligase, which helps to add SUMO onto several proteins and in tumor
progression [35, 36].
Cancer cells are highly proliferating cells, which demand large energy inputs, and mitochondrion is the predominant source of energy in cell. Therefore, maintaining mitochondrial health is of paramount significance in cancer cells, and several PQC members, including E3 ligases, help in achieving it via mitochondria-associated degradation (MAD) [37]. Proteins that are misfolded are often trafficked to the outer mitochondria membrane [38], where they are dealt with by Cdc48 (p97) [39]. Cdc48, along with other co-factors (HSP70 and HSC70), is known to be greatly enhanced in cancer owing to overwhelming proteotoxic stress and helps ubiquitinate and degrade the misfolded clients at mitochondria and ER membrane [40, 41]. Besides, Cdc48 can also eliminate the misfolded proteins at mitochondria by initiating the autophagic degradation of mitochondria with aid of Parkin [42, 43]. Autophagy, in general, is upregulated in cancer cells owing to deprived nutrient and hypoxia conditions. The key elements of autophagy initiation, which includes ULK1, help in phagophore formation, are greatly increased in cancerous cells [44]. Besides, ATG proteins also help in the phagophore formation and elongation and mediate the loading of LC3 (substrate receptor) [45]. LC3 is present in greater levels, helping in capture and selection of substrate, along with elongation and maturation of autophagosome in cancer [46, 47].
LC3 interacts with cargo receptor of autophagy i.e., p62, which is also known to be aberrantly increased in cancer conditions. The misfolded proteins are ubiquitinated by different E3 ligases, and these ubiquitinated misfolded proteins can interact with p62, which on the counterpart interacts with LC3 on membrane of autophagosome [48]. Finally, the substrate capture autophagosome then goes ahead and combines with the lysosome forming autophago-lysosome where the substrate demolition occurs by the lytic enzymes. The intersection and blending of the autophagosome with the lysosome are largely mediated by the critical protein Beclin1, which again is highly upregulated in cancers [49]. Autophagy can have double role in cancer cell as it can promote oncogenesis as well in many instances, it can inhibit cancer progression [50]. If we focus on its oncogenic nature, it can help in cell survival, inhibition of apoptosis, hypoxia response, and developing resistance to drugs in cancerous cells [45]. Overall, these homeostatic mechanisms, collectively understood as the protein quality control system are found to be greatly enhanced in cancer cells. These mechanisms help to take care of the widely unstable proteome and mediate the cell cycle deregulation, achieve immortalization, mediate metastasis and angiogenesis, and employ drug resistance. In the upcoming sections, we will thoroughly discuss these mechanisms in cancer cells.
How Does Ubiquitin Proteasome System Regulate the Tumor Cell Progression?
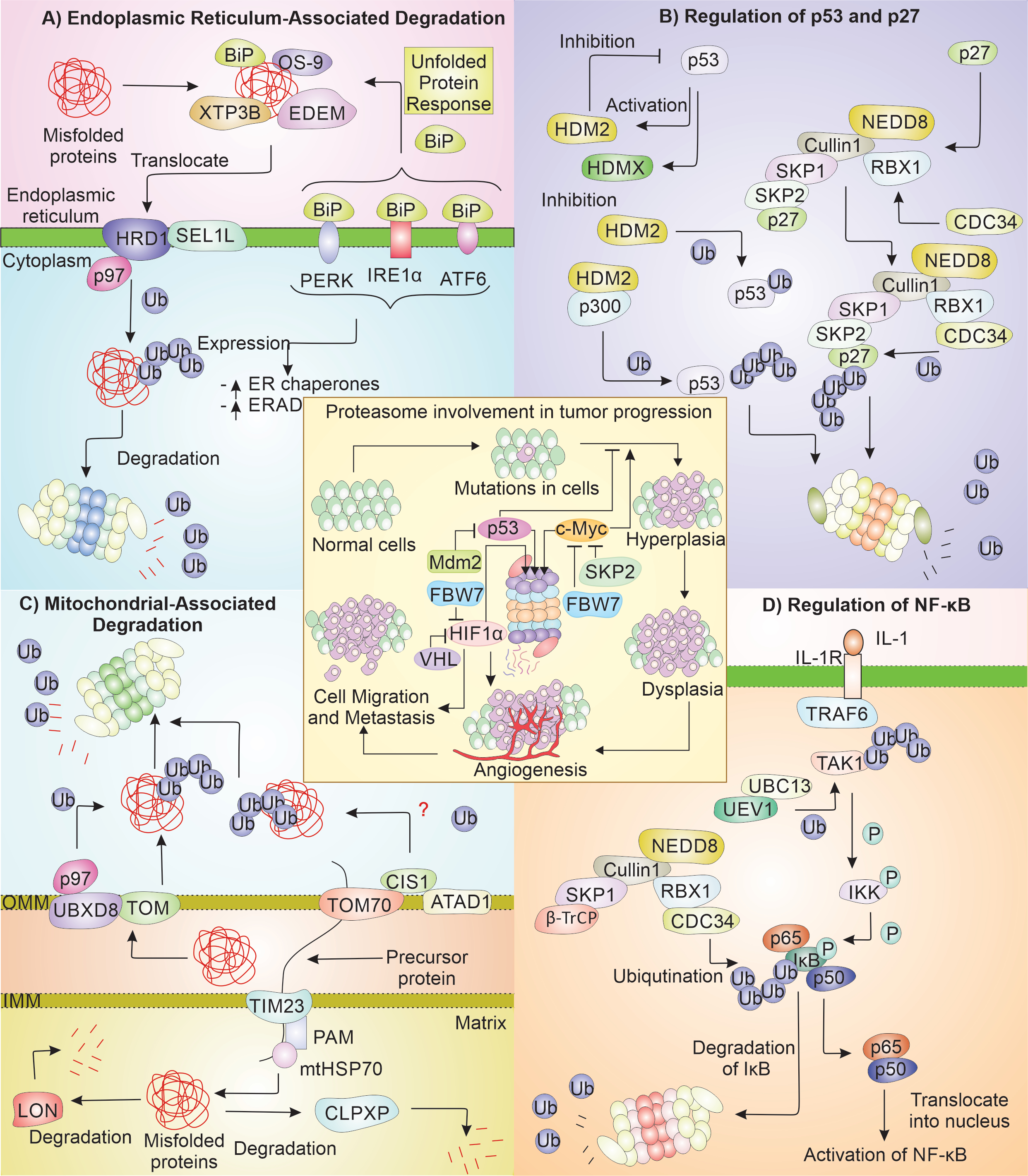
The UPS degrades most of the misfolded proteins present in the cells [51]. In a cell, UPS contributes to reducing the impaired proteins and re-modeling of the mitochondrial proteome during stress [52]. UPS also regulates the important proteins essential to cell-cycle progress like p53, p27 and the mitochondrial protein quality control [53, 54]. One out of three of the total proteins of the cell is folded in ER and moved to different cellular compartments. The maintenance of homeostasis in ER is done by two ER quality regulating machinery, ERAD and UPR [55] (Fig. 2).
Enhanced Cytoprotective Functions of Endoplasmic Reticulum Based Quality Control Systems and their Possible Implications in Cancer Cells
The ER is central to the translation as well as further modifications of the secretory proteins. Changes in the ER environment either by physiological or pathological stimuli can lead to protein aggregation, calcium depletion, and oxidative stress [56, 57]. ER quality control helps in maintaining the homeostasis aiding in survival and proliferation of cancerous cells [58]. Stress at ER triggers the UPR, which help the cancer cells to survive under stress conditions [55]. There are three pathways that control the protein quality control (PQC) of ER and maintain ER homeostasis - UPR, ERAD, and autophagy. ERAD directs improperly folded proteins present inside ER to get degraded by the proteasome. ERAD plays part in identifying misfolded proteins, their transportation across ER-cytosol, and finally, their ubiquitination directing them to proteasome [59]. Improperly folded proteins in ER are identified by BiP [60], OS-9 [61], XTP3B, and EDEM [62]. After recognition of the aberrantly folded protein, it is translocated into the cytoplasm and ubiquitinylated via HRD1 [63], in complex with Sel1L [64, 65]. UPR is also activated in cancer, which leads to the initiation of the PERK, IRE1α, and ATF6, which are ER stress sensors [66]. These sensors get activated when BiP detaches from their luminal part. Each ER sensor ultimately leads to the rise in the level of ER chaperones as well as increases the ERAD. The regulation of ER chaperones and the sensors of UPR have been reported as potential cancer therapies [67]. Recently, PERK-activated protein ATF4 has been found to increase by several folds in hypoxia environment in breast cancer tissue [68].
Importance of Ubiquitin-Proteasome System-Mediated Regulation of p53 and p27 in Establishing Tumorigenesis
p53 and p27 are the important proteins, which regulate the cell cycle. p53 is the essential tumor suppressor, with many cancer patients having defects in the p53 signaling [69]. p53 is a nuclear TF, which influences the transcription of various genes engaged in promoting cell death and arrest of cell cycle [70]. The proteasomal degradation maintains lower levels of p53 in regular physiological environment. But when DNA is damaged in cells irreversibly, p53 shows its pro-apoptotic function, removing damaged cells and thus prevent their further division. The p27 protein is a CDK inhibitor regulating the cell-division cycle advancement to the S phase from G1 [71]. p53 activity is vital for suppressing tumor, and its stability is affected by HDM2 and HDMX [72]. It has been noted that increase in HDM2 leads to a decrease in p53. The E3 ligase HDM2 alone can mono-ubiquitinate p53, which is not degraded by the proteasome instead is exported out of the nucleus, but when HDM2 is with p300, it polyubiquitinates the p53, which help in its degradation, thus supporting tumor growth [53]. p27 protein is the main controller of the cell cycle by controlling the CDK function, and reduction in its level via proteasome is required for the cell to enter the state of proliferation. The proteasomal degradation of p27 is mediated by NEDD8ylated SCFSkp2 complex (E3) consisting of Cullin1, SKP1, RBX1, and SKP2 [54, 73, 74]. It has been noted that many cancers have high levels of SKP2 and CDC34 (E2 enzyme) and reduced p27 [75, 76]. The reduced level of p27 is common among multiple cancers like ovarian and breast cancer [77].
Increased Mitochondrial-Associated Degradation Aids in Tumor Cell Proteostasis
The functioning of mitochondria is supported by UPS for the degradation of the misfolded proteins that are accumulated during stress conditions. TOM complex is involved in maintenance of protein quality along with UBXD8 (UBX2 in yeast) at outer mitochondrial membrane (OMM) [78]. UBX2 has a UBX domain, which can bind to the CDC48, and this protein also has affinity for ubiquitinated proteins [79, 80]. UBXD8 with p97 is engaged in degrading the mitochondrial proteins by targeting them to the proteasome, and they are upregulated in proliferating cancerous cells [81]. Many cancers show high expression of TOMM20 [82]. Another mitochondrial PQC mechanism, the mitoCPR degrades the incorrectly folded proteins on cytosolic face of OMM during protein import stress [83, 84]. CIS1 is a vital protein of this system, expression of which is very important for overcoming the compromised protein import. CIS1 interacts with TOM70, recruiting ATAD1 (AAA ATPase; MSP1 in yeast) to clear the proteins from the TOM by their degradation via proteasome [84, 85], while proteins that are misfolded in the matrix of the mitochondria are degraded by LON protease [86] and CLPXP protease [87-89]. LON protease has the vital function in tumor growth [86] and ClpXP protease levels has also shown to be elevated in tumors and hematologic malignancies [88, 89].
Proteasomal System Based Modulation of NF-κB in Cancer Biology
The TF NF-κB is essential in cell survival. It has subunits: Rel, p105/p50, p100/p52, p65, and RelB [90]. N-terminal RHD and C-terminal TAD occurs in p65, Rel, and RelB, while p52 and p50 have only RHD [91]. RHD is required in dimerization and DNA binding, while TAD is required for transcriptional activation [92]. NF-κB as p50-p65 dimer binds DNA. The NF-κB pathway is engaged in numerous kinds of tumors [93]. During cell homeostasis, NF-κB complex is stuck in the cytosol via the attachment to IκB, which inhibits NF-κB by preventing transfer to nucleus. On receiving signal on cell surface receptors like IL-1R and TNFR, by IL-1 or TNF, TRAF6 (E3) along with UBC13-UEV1 (E2) helps in attaching polyubiquitin chain to TAK1 [94]. The polyubiquitin chain also recruits IκB kinase, which is later activated by TAK1 kinase function [95]. The activated IκB kinase then phosphorylates IκB making it prone to ubiquitination by SCFβ-TrCP complex, and directed to proteasome for demolition [96]. The degradation of IκB frees NF-κB from its association in the cytosol, to move to nucleus, initiating its signaling pathway [97]. NF-κB affects the tumor progression and development by activation of excessive innate immunity and promotes abnormal cell growth [98]. NF-κB pathway is altered in hematopoietic and tumor malignancy, promoting tumor cell division and survival [99, 100]. Contrary to this, NF-κB is understood to reduce the progress of
tumor [101].
Author Contributions
A.R.D, P.K, S.K, Y.A.J, and S.M.P performed the figures design framework and perform the writing of first draft. S·S, A.K, AW.K, perform critical observations of manuscript, verified and the finalize draft in line with concept of manuscript. A.M contributed to the overall design of concept in figures formation, formulate and analyze the contents and perform the final writing of the manuscript. All authors discussed the results and contributed to the final manuscript.
Funding
The presented work was supported by the Science & Engineering Research Board (SERB), Department of Science & Technology, Government of India grant to (AM) EMR/2016/000716 and from BRNS grant to (AM) 54/14/16/2020-BRNS-Government of India.
The authors have no conflicts of interest to declare.
| 1 Faguet GB: A brief history of cancer: age‐old milestones underlying our current knowledge database. Int J Cancer 2015;136:2022-2036. https://doi.org/10.1002/ijc.29134 |
||||
| 2 Di Lonardo A, Nasi S, Pulciani S: Cancer: we should not forget the past. J Cancer 2015;6:29. https://doi.org/10.7150/jca.10336 |
||||
| 3 Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011;144:646-674. https://doi.org/10.1016/j.cell.2011.02.013 |
||||
| 4 Mitrus I, Bryndza E, Sochanik A, Szala S: Evolving models of tumor origin and progression. Tumour Biol 2012;33:911-917. https://doi.org/10.1007/s13277-012-0389-0 |
||||
| 5 Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Ulrich Hartl F: Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 2013;82:323-355. https://doi.org/10.1146/annurev-biochem-060208-092442 |
||||
| 6 Albakova Z, Armeev GA, Kanevskiy LM, Kovalenko EI, Sapozhnikov AM: HSP70 Multi-Functionality in Cancer. Cells 2020;9:587. https://doi.org/10.3390/cells9030587 |
||||
| 7 Jafari A, Rezaei-Tavirani M, Farhadihosseinabadi B, Taranejoo S, Zali H: HSP90 and Co-chaperones: Impact on Tumor Progression and Prospects for Molecular-Targeted Cancer Therapy. Cancer Invest 2020;38:310-328. https://doi.org/10.1080/07357907.2020.1752227 |
||||
| 8 Fan W, Fan SS, Feng J, Xiao D, Fan S, Luo J: Elevated expression of HSP10 protein inhibits apoptosis and associates with poor prognosis of astrocytoma. PLoS One 2017;12:e0185563. https://doi.org/10.1371/journal.pone.0185563 |
||||
| 9 Fucarino A, Pitruzzella A: Role of HSP60/HSP10 in Lung Cancer: Simple Biomarkers or Leading Actors? J Oncol 2020;2020:4701868. https://doi.org/10.1155/2020/4701868 |
||||
| 10 Zhou C, Sun H, Zheng C, Gao J, Fu Q, Hu N, Shao X, Zhou Y, Xiong J, Nie K: Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis 2018;9:1-14. https://doi.org/10.1038/s41419-017-0196-z |
||||
| 11 Choi SK, Kam H, Kim KY, Park SI, Lee YS: Targeting heat shock protein 27 in cancer: a druggable target for cancer treatment? Cancers 2019;11:1195. https://doi.org/10.3390/cancers11081195 |
||||
| 12 Sheng B, Qi C, Liu B, Lin Y, Fu T, Zeng Q: Increased HSP27 correlates with malignant biological behavior of non-small cell lung cancer and predicts patient's survival. Sci Rep 2017;7:1-12. https://doi.org/10.1038/s41598-017-13956-2 |
||||
| 13 Yadav RK, Chae SW, Kim HR, Chae HJ: Endoplasmic reticulum stress and cancer. J Cancer Prev 2014;19:75. https://doi.org/10.15430/JCP.2014.19.2.75 |
||||
| 14 Cook KL, Clarke R: Role of GRP78 in promoting therapeutic-resistant breast cancer. Future Med Chem 2015;7:1529-1534. https://doi.org/10.4155/fmc.15.80 |
||||
| 15 Dey S, Tameire F, Koumenis C: PERK-ing up autophagy during MYC-induced tumorigenesis. Autophagy 2013;9:612-614. https://doi.org/10.4161/auto.23486 |
||||
| 16 Walczak A, Gradzik K, Kabzinski J, Przybylowska-Sygut K, Majsterek I: The role of the ER-induced UPR pathway and the efficacy of its inhibitors and inducers in the inhibition of tumor progression. Oxid Med Cell Longev 2019;2019:5729710. https://doi.org/10.1155/2019/5729710 |
||||
| 17 Deng P, Haynes CM: Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol 2017;47:43-49. https://doi.org/10.1016/j.semcancer.2017.05.002 |
||||
| 18 Kim H, Bhattacharya A, Qi L: Endoplasmic reticulum quality control in cancer: Friend or foe. Semin Cancer Biol 2015;33:25-33. https://doi.org/10.1016/j.semcancer.2015.02.003 |
||||
| 19 Biunno I, Cattaneo M, Orlandi R, Canton C, Biagiotti L, Ferrero S, Barberis M, Pupa SM, Scarpa A, Ménard S: SEL1L a multifaceted protein playing a role in tumor progression. J Cell Physiol 2006;208:23-38. https://doi.org/10.1002/jcp.20574 |
||||
| 20 Tan X, He X, Fan Z: Upregulation of HRD1 promotes cell migration and invasion in colon cancer. Mol Cell Biochem 2019;454:1-9. https://doi.org/10.1007/s11010-018-3447-0 |
||||
| 21 Onishi Y, Tsukada K, Yokota J, Raz A: Overexpression of autocrine motility factor receptor (AMFR) in NIH3T3 fibroblasts induces cell transformation. Clin Exp Metastasis 2003;20:51-58. https://doi.org/10.1023/A:1022594503657 |
||||
| 22 Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, Chen B, Veenstra T, Hewitt SM, Helman LJ, Khanna C: The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med 2007;13:1504-1509. https://doi.org/10.1038/nm1686 |
||||
| 23 Morozov AV, Karpov VL: Proteasomes and several aspects of their heterogeneity relevant to cancer. Front Oncol 2019;9:761. https://doi.org/10.3389/fonc.2019.00761 |
||||
| 24 Ciechanover A, Brundin P: The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 2003;40:427-446. https://doi.org/10.1016/S0896-6273(03)00606-8 |
||||
| 25 Tokgöz Z, Bohnsack RN, Haas AL: Pleiotropic effects of ATP·Mg2+ binding in the catalytic cycle of ubiquitin-activating enzyme. J Biol Chem 2006;281:14729-14737. https://doi.org/10.1074/jbc.M513562200 |
||||
| 26 Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM: RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci U S A 1999;96:11364-11369. https://doi.org/10.1073/pnas.96.20.11364 |
||||
| 27 Berndsen CE, Wolberger C: New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 2014;21:301. https://doi.org/10.1038/nsmb.2780 |
||||
| 28 Murata S, Minami Y, Minami M, Chiba T, Tanaka K: CHIP is a chaperone‐dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2001;2:1133-1138. https://doi.org/10.1093/embo-reports/kve246 |
||||
| 29 Andrews P, He Y, Xiong Y: Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene 2006;25:4534-4548. https://doi.org/10.1038/sj.onc.1209490 |
||||
| 30 Dornan D, Bheddah S, Newton K, Ince W, Frantz GD, Dowd P, Koeppen H, Dixit VM, French DM: COP1, the negative regulator of p53, is overexpressed in breast and ovarian adenocarcinomas. Cancer Res 2004;64:7226-7230. https://doi.org/10.1158/0008-5472.CAN-04-2601 |
||||
| 31 Hou H, Sun D, Zhang X: The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int 2019;19:1-8. https://doi.org/10.1186/s12935-019-0937-4 |
||||
| 32 Ka WH, Cho SK, Chun BN, Byun SY, Ahn JC: The ubiquitin ligase COP1 regulates cell cycle and apoptosis by affecting p53 function in human breast cancer cell lines. Breast Cancer 2018;25:529-538. https://doi.org/10.1007/s12282-018-0849-5 |
||||
| 33 Gamell C, Bandilovska I, Gulati T, Kogan A, Lim SC, Kovacevic Z, Takano EA, Timpone C, Agupitan AD, Litchfield C: E6AP promotes a metastatic phenotype in prostate cancer. iScience 2019;22:1-15. https://doi.org/10.1016/j.isci.2019.10.065 |
||||
| 34 Eide PW, Cekaite L, Danielsen SA, Eilertsen IA, Kjenseth A, Fykerud TA, Ågesen TH, Bruun J, Rivedal E, Lothe RA: NEDD4 is overexpressed in colorectal cancer and promotes colonic cell growth independently of the PI3K/PTEN/AKT pathway. Cell Signal 2013;25:12-18. https://doi.org/10.1016/j.cellsig.2012.08.012 |
||||
| 35 Chen C, Matesic LE: The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev 2007;26:587. https://doi.org/10.1007/s10555-007-9091-x |
||||
| 36 Rabellino A, Andreani C, Scaglioni PP: The role of PIAS SUMO E3-ligases in cancer. Cancer Res 2017;77:1542-1547. https://doi.org/10.1158/0008-5472.CAN-16-2958 |
||||
| 37 Liao PC, Wolken DMA, Serrano E, Srivastava P, Pon LA: Mitochondria-associated degradation pathway (MAD) function beyond the outer membrane. Cell Rep 2020;32:107902. https://doi.org/10.1016/j.celrep.2020.107902 |
||||
| 38 Biel TG, Aryal B, Gerber MH, Trevino JG, Mizuno N, Rao VA: Mitochondrial dysfunction generates aggregates that resist lysosomal degradation in human breast cancer cells. Cell Death Dis 2020;11:1-15. https://doi.org/10.1038/s41419-020-2658-y |
||||
| 39 Escobar-Henriques M, Anton V: Mitochondrial Surveillance by Cdc48/p97: MAD vs. Membrane Fusion. Int J Mol Sci 2020;21:6841. https://doi.org/10.3390/ijms21186841 |
||||
| 40 Thoms S: Cdc48 can distinguish between native and non-native proteins in the absence of cofactors. FEBS Lett 2002;520:107-110. https://doi.org/10.1016/S0014-5793(02)02777-1 |
||||
| 41 Vekaria PH, Home T, Weir S, Schoenen FJ, Rao R: Targeting p97 to disrupt protein homeostasis in cancer. Front Oncol 2016;6:181. https://doi.org/10.3389/fonc.2016.00181 |
||||
| 42 da Silva-Camargo CCV, Baldin RKS, Polli NLC, Agostinho AP, Olandosk M, de Noronha L, Sotomaior VS: Parkin protein expression and its impact on survival of patients with advanced colorectal cancer. Cancer Biol Med 2018;15:61. | ||||
| 43 Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ: Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010;191:1367-1380. https://doi.org/10.1083/jcb.201007013 |
||||
| 44 Lu J, Zhu L, Zheng LP, Cui Q, Zhu HH, Zhao H, Shen ZJ, Dong HY, Chen SS, Wu WZ: Overexpression of ULK1 represents a potential diagnostic marker for clear cell renal carcinoma and the antitumor effects of SBI-0206965. EBioMedicine 2018;34:85-93. https://doi.org/10.1016/j.ebiom.2018.07.034 |
||||
| 45 Li X, He S, Ma B: Autophagy and autophagy-related proteins in cancer. Mol Cancer 2020;19:12. https://doi.org/10.1186/s12943-020-1138-4 |
||||
| 46 Tanida I, Ueno T, Kominami E: LC3 and Autophagy. Methods Mol Biol 2008;445:77-88. https://doi.org/10.1007/978-1-59745-157-4_4 |
||||
| 47 Yoshioka A, Miyata H, Doki Y, Yamasaki M, Sohma I, Gotoh K, Takiguchi S, Fujiwara Y, Uchiyama Y, Monden M: LC3, an autophagosome marker, is highly expressed in gastrointestinal cancers. Int J Oncol 2008;33:461-468. | ||||
| 48 Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, Johansen T: p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131-24145. https://doi.org/10.1074/jbc.M702824200 |
||||
| 49 Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H: Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013;32:2261-2272. https://doi.org/10.1038/onc.2012.252 |
||||
| 50 Levy JMM, Towers CG, Thorburn A: Targeting autophagy in cancer. Nat Rev Cancer 2017;17:528-542. https://doi.org/10.1038/nrc.2017.53 |
||||
| 51 Lee DH, Goldberg AL: Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol 1998;8:397-403. https://doi.org/10.1016/S0962-8924(98)01346-4 |
||||
| 52 Bragoszewski P, Turek M, Chacinska A: Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol 2017;7:170007. https://doi.org/10.1098/rsob.170007 |
||||
| 53 Moll UM, Petrenko O: The MDM2-p53 interaction. Mol Cancer Res 2003;1:1001-1008. | ||||
| 54 Bloom J, Pagano M: Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol 2003;13:41-47. https://doi.org/10.1016/S1044-579X(02)00098-6 |
||||
| 55 Tsai YC, Weissman AM: The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010;1:764-778. https://doi.org/10.1177/1947601910383011 |
||||
| 56 Cao SS, Kaufman RJ: Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 2014;21:396-413. https://doi.org/10.1089/ars.2014.5851 |
||||
| 57 Hetz C: The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012;13:89-102. https://doi.org/10.1038/nrm3270 |
||||
| 58 Martinon F: Targeting endoplasmic reticulum signaling pathways in cancer. Acta Oncol 2012;51:822-830. https://doi.org/10.3109/0284186X.2012.689113 |
||||
| 59 Meusser B, Hirsch C, Jarosch E, Sommer T: ERAD: the long road to destruction. Nat Cell Biol 2005;7:766-772. https://doi.org/10.1038/ncb0805-766 |
||||
| 60 Li J, Lee AS: Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med 2006;6:45-54. https://doi.org/10.2174/156652406775574523 |
||||
| 61 Christianson JC, Shaler TA, Tyler RE, Kopito RR: OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol 2008;10:272-282. https://doi.org/10.1038/ncb1689 |
||||
| 62 Molinari M, Calanca V, Galli C, Lucca P, Paganetti P: Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003;299:1397-1400. https://doi.org/10.1126/science.1079474 |
||||
| 63 Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY: Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 2001;3:24-29. https://doi.org/10.1038/35050524 |
||||
| 64 Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonca N, Liu X, Locasale JW, Simpson KW, Duhamel GE, Kersten S, Yates JR 3rd, Long Q, Qi L: Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A 2014;111:E582-591. https://doi.org/10.1073/pnas.1318114111 |
||||
| 65 Mueller B, Klemm EJ, Spooner E, Claessen JH, Ploegh HL: SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci U S A 2008;105:12325-12330. https://doi.org/10.1073/pnas.0805371105 |
||||
| 66 Ranatunga S, Tang CH, Kang CW, Kriss CL, Kloppenburg BJ, Hu CC, Del Valle JR: Synthesis of novel tricyclic chromenone-based inhibitors of IRE-1 RNase activity. J Med Chem 2014;57:4289-4301. https://doi.org/10.1021/jm5002452 |
||||
| 67 Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, Polverini PJ: The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res 2012;72:5396-5406. https://doi.org/10.1158/0008-5472.CAN-12-0474 |
||||
| 68 Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC, Span PN: Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res 2013;15:R2. https://doi.org/10.1186/bcr3373 |
||||
| 69 Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP: Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009;9:862-873. https://doi.org/10.1038/nrc2763 |
||||
| 70 Vousden KH, Lu X: Live or let die: the cell's response to p53. Nat Rev Cancer 2002;2:594-604. https://doi.org/10.1038/nrc864 |
||||
| 71 Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI: A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci U S A 1994;91:5291-5295. https://doi.org/10.1073/pnas.91.12.5291 |
||||
| 72 Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP: Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996;274:948-953. https://doi.org/10.1126/science.274.5289.948 |
||||
| 73 Carrano AC, Eytan E, Hershko A, Pagano M: SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999;1:193-199. https://doi.org/10.1038/12013 |
||||
| 74 Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP: Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 2002;416:703-709. https://doi.org/10.1038/416703a |
||||
| 75 Chiarle R, Fan Y, Piva R, Boggino H, Skolnik J, Novero D, Palestro G, De Wolf-Peeters C, Chilosi M, Pagano M, Inghirami G: S-phase kinase-associated protein 2 expression in non-Hodgkin's lymphoma inversely correlates with p27 expression and defines cells in S phase. Am J Pathol 2002;160:1457-1466. https://doi.org/10.1016/S0002-9440(10)62571-0 |
||||
| 76 Chauhan D, Li G, Hideshima T, Podar K, Shringarpure R, Mitsiades C, Munshi N, Yew PR, Anderson KC: Blockade of ubiquitin-conjugating enzyme CDC34 enhances anti-myeloma activity of Bortezomib/Proteasome inhibitor PS-341. Oncogene 2004;23:3597-3602. https://doi.org/10.1038/sj.onc.1207458 |
||||
| 77 Chu IM, Hengst L, Slingerland JM: The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008;8:253-267. https://doi.org/10.1038/nrc2347 |
||||
| 78 Martensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, Becker T: Mitochondrial protein translocation-associated degradation. Nature 2019;569:679-683. https://doi.org/10.1038/s41586-019-1227-y |
||||
| 79 Metzger MB, Scales JL, Dunklebarger MF, Loncarek J, Weissman AM: A protein quality control pathway at the mitochondrial outer membrane. Elife 2020;9:e51065. https://doi.org/10.7554/eLife.51065 |
||||
| 80 Schuberth C, Buchberger A: Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol 2005;7:999-1006. https://doi.org/10.1038/ncb1299 |
||||
| 81 Haines DS: p97-containing complexes in proliferation control and cancer: emerging culprits or guilt by association? Genes Cancer 2010;1:753-763. https://doi.org/10.1177/1947601910381381 |
||||
| 82 Park SH, Lee AR, Choi K, Joung S, Yoon JB, Kim S: TOMM20 as a potential therapeutic target of colorectal cancer. BMB Rep 2019;52:712-717. https://doi.org/10.5483/BMBRep.2019.52.12.249 |
||||
| 83 Boos F, Kramer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zoller E, van der Laan M, Savitski MM, Benes V, Herrmann JM: Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat Cell Biol 2019;21:442-451. https://doi.org/10.1038/s41556-019-0294-5 |
||||
| 84 Weidberg H, Amon A: MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 2018;360:eaan4146. https://doi.org/10.1126/science.aan4146 |
||||
| 85 Basch M, Wagner M, Rolland S, Carbonell A, Zeng R, Khosravi S, Schmidt A, Aftab W, Imhof A, Wagener J, Conradt B, Wagener N: Msp1 cooperates with the proteasome for extraction of arrested mitochondrial import intermediates. Mol Biol Cell 2020;31:753-767. https://doi.org/10.1091/mbc.E19-06-0329 |
||||
| 86 Quiros PM, Barcena C, Lopez-Otin C: Lon protease: A key enzyme controlling mitochondrial bioenergetics in cancer. Mol Cell Oncol 2014;1:e968505. https://doi.org/10.4161/23723548.2014.968505 |
||||
| 87 Koppen M, Langer T: Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit Rev Biochem Mol Biol 2007;42:221-242. https://doi.org/10.1080/10409230701380452 |
||||
| 88 Seo JH, Rivadeneira DB, Caino MC, Chae YC, Speicher DW, Tang HY, Vaira V, Bosari S, Palleschi A, Rampini P, Kossenkov AV, Languino LR, Altieri DC: The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol 2016;14:e1002507. https://doi.org/10.1371/journal.pbio.1002507 |
||||
| 89 Nouri K, Feng Y, Schimmer AD: Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy. Cell Death Dis 2020;11:841. https://doi.org/10.1038/s41419-020-03062-z |
||||
| 90 Kim JS, Kim EJ, Kim HS, Kurie JM, Ahn YH: MKK4 activates non-canonical NFkappaB signaling by promoting NFkappaB2-p100 processing. Biochem Biophys Res Commun 2017;491:337-342. https://doi.org/10.1016/j.bbrc.2017.07.099 |
||||
| 91 Keogh CE, Scholz CC, Rodriguez J, Selfridge AC, von Kriegsheim A, Cummins EP: Carbon dioxide-dependent regulation of NF-kappaB family members RelB and p100 gives molecular insight into CO2-dependent immune regulation. J Biol Chem 2017;292:11561-11571. https://doi.org/10.1074/jbc.M116.755090 |
||||
| 92 Isoda K, Roth S, Nusslein-Volhard C: The functional domains of the Drosophila morphogen dorsal: evidence from the analysis of mutants. Genes Dev 1992;6:619-630. https://doi.org/10.1101/gad.6.4.619 |
||||
| 93 Prasad S, Ravindran J, Aggarwal BB: NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem 2010;336:25-37. https://doi.org/10.1007/s11010-009-0267-2 |
||||
| 94 Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, Zheng L, Rich RL, Campos AD, Myszka DG, Lenardo MJ, Darnay BG, Wu H: E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol 2009;16:658-666. https://doi.org/10.1038/nsmb.1605 |
||||
| 95 Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ: TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001;412:346-351. https://doi.org/10.1038/35085597 |
||||
| 96 Shambharkar PB, Blonska M, Pappu BP, Li H, You Y, Sakurai H, Darnay BG, Hara H, Penninger J, Lin X: Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. EMBO J 2007;26:1794-1805. https://doi.org/10.1038/sj.emboj.7601622 |
||||
| 97 Iwai K: Diverse ubiquitin signaling in NF-kappaB activation. Trends Cell Biol 2012;22:355-364. https://doi.org/10.1016/j.tcb.2012.04.001 |
||||
| 98 Grivennikov SI, Greten FR, Karin M: Immunity, inflammation, and cancer. Cell 2010;140:883-899. https://doi.org/10.1016/j.cell.2010.01.025 |
||||
| 99 Sau A, Lau R, Cabrita MA, Nolan E, Crooks PA, Visvader JE, Pratt MA: Persistent Activation of NF-kappaB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA Damage. Cell Stem Cell 2016;19:52-65. https://doi.org/10.1016/j.stem.2016.05.003 |
||||
| 100 Salazar L, Kashiwada T, Krejci P, Meyer AN, Casale M, Hallowell M, Wilcox WR, Donoghue DJ, Thompson LM: Fibroblast growth factor receptor 3 interacts with and activates TGFbeta-activated kinase 1 tyrosine phosphorylation and NFkappaB signaling in multiple myeloma and bladder cancer. PLoS One 2014;9:e86470. https://doi.org/10.1371/journal.pone.0086470 |
||||
| 101 Liu F, Bardhan K, Yang D, Thangaraju M, Ganapathy V, Waller JL, Liles GB, Lee JR, Liu K: NF-kappaB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression. J Biol Chem 2012;287:25530-25540. https://doi.org/10.1074/jbc.M112.356279 |
||||
| 102 Zheng N, Shabek N: Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 2017;86:129-157. https://doi.org/10.1146/annurev-biochem-060815-014922 |
||||
| 103 Akutsu M, Dikic I, Bremm A: Ubiquitin chain diversity at a glance. J Cell Sci 2016;129:875-880. https://doi.org/10.1242/jcs.183954 |
||||
| 104 Kleiger G, Mayor T: Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol 2014;24:352-359. https://doi.org/10.1016/j.tcb.2013.12.003 |
||||
| 105 Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM: Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 2007;67:9472-9481. https://doi.org/10.1158/0008-5472.CAN-07-0568 |
||||
| 106 Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, Huck J, Shi J, Gavin J, Brownell J, Yang Y, Stringer B, Griffin R, Bruzzese F, Soucy T, Duffy J, Rabino C, Riceberg J, Hoar K, Lublinsky A, et al.: A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 2018;24:186-193. https://doi.org/10.1038/nm.4474 |
||||
| 107 An H, Statsyuk AV: An inhibitor of ubiquitin conjugation and aggresome formation. Chem Sci 2015;6:5235-5245. https://doi.org/10.1039/C5SC01351H |
||||
| 108 Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, et al.: An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009;458:732-736. https://doi.org/10.1038/nature07884 |
||||
| 109 Muraoka H, Yoshimura C, Kawabata R, Tsuji S, Hashimoto A, Ochiiwa H, Nakagawa F, Fujioka Y, Matsuo K, Ohkubo S: Activity of TAS4464, a novel NEDD8 activating enzyme E1 inhibitor, against multiple myeloma via inactivation of nuclear factor kappaB pathways. Cancer Sci 2019;110:3802-3810. https://doi.org/10.1111/cas.14209 |
||||
| 110 Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V, Neculai D, Chou YC, Ogunjimi A, Al-Hakim A, Varelas X, Koszela J, Wasney GA, Vedadi M, Dhe-Paganon S, Cox S, Xu S, Lopez-Girona A, Mercurio F, Wrana J, et al.: An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 2011;145:1075-1087. https://doi.org/10.1016/j.cell.2011.05.039 |
||||
| 111 Hodge CD, Edwards RA, Markin CJ, McDonald D, Pulvino M, Huen MS, Zhao J, Spyracopoulos L, Hendzel MJ, Glover JN: Covalent Inhibition of Ubc13 Affects Ubiquitin Signaling and Reveals Active Site Elements Important for Targeting. ACS Chem Biol 2015;10:1718-1728. https://doi.org/10.1021/acschembio.5b00222 |
||||
| 112 Tsukamoto S, Takeuchi T, Rotinsulu H, Mangindaan RE, van Soest RW, Ukai K, Kobayashi H, Namikoshi M, Ohta T, Yokosawa H: Leucettamol A: a new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg Med Chem Lett 2008;18:6319-6320. https://doi.org/10.1016/j.bmcl.2008.10.110 |
||||
| 113 Ushiyama S, Umaoka H, Kato H, Suwa Y, Morioka H, Rotinsulu H, Losung F, Mangindaan RE, de Voogd NJ, Yokosawa H, Tsukamoto S: Manadosterols A and B, sulfonated sterol dimers inhibiting the Ubc13-Uev1A interaction, isolated from the marine sponge Lissodendryx fibrosa. J Nat Prod 2012;75:1495-1499. https://doi.org/10.1021/np300352u |
||||
| 114 Giaccia AJ, Kastan MB: The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998;12:2973-2983. https://doi.org/10.1101/gad.12.19.2973 |
||||
| 115 Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA: In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004;303:844-848. https://doi.org/10.1126/science.1092472 |
||||
| 116 Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, Liu JJ, Zhao C, Glenn K, Wen Y, Tovar C, Packman K, Vassilev L, Graves B: Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med Chem Lett 2013;4:466-469. https://doi.org/10.1021/ml4000657 |
||||
| 117 Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, Kirschbaum M, Iyer SP, Ruvolo V, Gonzalez GM, Huang X, Chen G, Graves B, Blotner S, Bridge P, Jukofsky L, et al.: Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res 2016;22:868-876. https://doi.org/10.1158/1078-0432.CCR-15-0481 |
||||
| 118 Canon J, Osgood T, Olson SH, Saiki AY, Robertson R, Yu D, Eksterowicz J, Ye Q, Jin L, Chen A, Zhou J, Cordover D, Kaufman S, Kendall R, Oliner JD, Coxon A, Radinsky R: The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol Cancer Ther 2015;14:649-658. https://doi.org/10.1158/1535-7163.MCT-14-0710 |
||||
| 119 Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, Soria JC, Chawla S, de Weger V, Wagner AJ, Siegel D, De Vos F, Rasmussen E, Henary HA: Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs 2020;38:831-843. https://doi.org/10.1007/s10637-019-00840-1 |
||||
| 120 Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, et al.: Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A 2008;105:3933-3938. https://doi.org/10.1073/pnas.0708917105 |
||||
| 121 Canner JA, Sobo M, Ball S, Hutzen B, DeAngelis S, Willis W, Studebaker AW, Ding K, Wang S, Yang D, Lin J: MI-63: a novel small-molecule inhibitor targets MDM2 and induces apoptosis in embryonal and alveolar rhabdomyosarcoma cells with wild-type p53. Br J Cancer 2009;101:774-781. https://doi.org/10.1038/sj.bjc.6605199 |
||||
| 122 Herman AG, Hayano M, Poyurovsky MV, Shimada K, Skouta R, Prives C, Stockwell BR: Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov 2011;1:312-325. https://doi.org/10.1158/2159-8290.CD-11-0104 |
||||
| 123 Wang H, Ma X, Ren S, Buolamwini JK, Yan C: A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther 2011;10:69-79. https://doi.org/10.1158/1535-7163.MCT-10-0581 |
||||
| 124 Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, Hathaway N, Dimova N, Cuny GD, King RW: Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010;18:382-395. https://doi.org/10.1016/j.ccr.2010.08.010 |
||||
| 125 Sackton KL, Dimova N, Zeng X, Tian W, Zhang M, Sackton TB, Meaders J, Pfaff KL, Sigoillot F, Yu H, Luo X, King RW: Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014;514:646-649. https://doi.org/10.1038/nature13660 |
||||
| 126 Richeson KV, Bodrug T, Sackton KL, Yamaguchi M, Paulo JA, Gygi SP, Schulman BA, Brown NG, King RW: Paradoxical mitotic exit induced by a small molecule inhibitor of APC/C(Cdc20). Nat Chem Biol 2020;16:546-555. https://doi.org/10.1038/s41589-020-0495-z |
||||
| 127 Blees JS, Bokesch HR, Rubsamen D, Schulz K, Milke L, Bajer MM, Gustafson KR, Henrich CJ, McMahon JB, Colburn NH, Schmid T, Brune B: Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase beta-TrCP1. PLoS One 2012;7:e46567. https://doi.org/10.1371/journal.pone.0046567 |
||||
| 128 Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, Schlossman R, Podar K, Munshi NC, Mitsiades N, Anderson KC: Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003;101:1530-1534. https://doi.org/10.1182/blood-2002-08-2543 |
||||
| 129 Shirley M: Ixazomib: First Global Approval. Drugs 2016;76:405-411. https://doi.org/10.1007/s40265-016-0548-5 |
||||
| 130 Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ: Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 2009;113:4667-4676. https://doi.org/10.1182/blood-2008-07-171637 |
||||
| 131 Zhou Y, Liu X, Xue J, Liu L, Liang T, Li W, Yang X, Hou X, Fang H: Discovery of Peptide Boronate Derivatives as Histone Deacetylase and Proteasome Dual Inhibitors for Overcoming Bortezomib Resistance of Multiple Myeloma. J Med Chem 2020;63:4701-4715. https://doi.org/10.1021/acs.jmedchem.9b02161 |
||||
| 132 Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H: Covalent modification of the active site threonine of proteasomal beta subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc Natl Acad Sci U S A 1997;94:6629-6634. https://doi.org/10.1073/pnas.94.13.6629 |
||||
| 133 Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP: From bortezomib to other inhibitors of the proteasome and beyond. Curr Pharm Des 2013;19:4025-4038. https://doi.org/10.2174/1381612811319220012 |
||||
| 134 Liu N, Li X, Huang H, Zhao C, Liao S, Yang C, Liu S, Song W, Lu X, Lan X, Chen X, Yi S, Xu L, Jiang L, Zhao C, Dong X, Zhou P, Li S, Wang S, Shi X, et al.: Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget 2014;5:5453-5471. https://doi.org/10.18632/oncotarget.2113 |
||||
| 135 Yang H, Chen D, Cui QC, Yuan X, Dou QP: Celastrol, a triterpene extracted from the Chinese "Thunder of God Vine," is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 2006;66:4758-4765. https://doi.org/10.1158/0008-5472.CAN-05-4529 |
||||
| 136 Contreras L, Calderon RI, Varela-Ramirez A, Zhang HY, Quan Y, Das U, Dimmock JR, Skouta R, Aguilera RJ: Induction of apoptosis via proteasome inhibition in leukemia/lymphoma cells by two potent piperidones. Cell Oncol (Dordr) 2018;41:623-636. https://doi.org/10.1007/s13402-018-0397-1 |
||||
| 137 Nam S, Smith DM, Dou QP: Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem 2001;276:13322-13330. https://doi.org/10.1074/jbc.M004209200 |
||||
| 138 Wang M, Zhang Y, Wang T, Zhang J, Zhou Z, Sun Y, Wang S, Shi Y, Luan X, Zhang Y, Wang Y, Wang Y, Zou Z, Kang L, Liu H: The USP7 Inhibitor P5091 Induces Cell Death in Ovarian Cancers with Different P53 Status. Cell Physiol Biochem 2017;43:1755-1766. https://doi.org/10.1159/000484062 |
||||
| 139 Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, Weinstock J, Kingsbury WD, Hideshima T, Shah PK, Minvielle S, Altun M, Kessler BM, Orlowski R, Richardson P, Munshi N, et al.: A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012;22:345-358. https://doi.org/10.1016/j.ccr.2012.08.007 |
||||
| 140 Nicholson B, Leach CA, Goldenberg SJ, Francis DM, Kodrasov MP, Tian X, Shanks J, Sterner DE, Bernal A, Mattern MR, Wilkinson KD, Butt TR: Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci 2008;17:1035-1043. https://doi.org/10.1110/ps.083450408 |
||||
| 141 Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C: Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res 2006;66:9235-9244. https://doi.org/10.1158/0008-5472.CAN-06-0702 |
||||
| 142 Zhang X, Pellegrini P, Saei AA, Hillert EK, Mazurkiewicz M, Olofsson MH, Zubarev RA, D'Arcy P, Linder S: The deubiquitinase inhibitor b-AP15 induces strong proteotoxic stress and mitochondrial damage. Biochem Pharmacol 2018;156:291-301. https://doi.org/10.1016/j.bcp.2018.08.039 |
||||
| 143 Tian Z, D'Arcy P, Wang X, Ray A, Tai YT, Hu Y, Carrasco RD, Richardson P, Linder S, Chauhan D, Anderson KC: A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014;123:706-716. https://doi.org/10.1182/blood-2013-05-500033 |
||||
| 144 Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, Bossler F, Khan AC, Linder S, D'Arcy P: The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci Rep 2016;6:26979. https://doi.org/10.1038/srep30667 |
||||
| 145 Chang HY, Yang X: Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev 2000;64:821-846. https://doi.org/10.1128/MMBR.64.4.821-846.2000 |
||||
| 146 Deshmukh M, Kuida K, Johnson EM, Jr.: Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol 2000;150:131-143. https://doi.org/10.1083/jcb.150.1.131 |
||||
| 147 Gama V, Swahari V, Schafer J, Kole AJ, Evans A, Huang Y, Cliffe A, Golitz B, Sciaky N, Pei XH, Xiong Y, Deshmukh M: The E3 ligase PARC mediates the degradation of cytosolic cytochrome c to promote survival in neurons and cancer cells. Sci Signal 2014;7:ra67. https://doi.org/10.1126/scisignal.2005309 |
||||
| 148 Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD: Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002;111:331-342. https://doi.org/10.1016/S0092-8674(02)01036-X |
||||
| 149 Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH: Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 2009;36:487-499. https://doi.org/10.1016/j.molcel.2009.09.030 |
||||
| 150 Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X: Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479-489. https://doi.org/10.1016/S0092-8674(00)80434-1 |
||||
| 151 Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ: Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 1999;144:281-292. https://doi.org/10.1083/jcb.144.2.281 |
||||
| 152 Jin Z, El-Deiry WS: Overview of cell death signaling pathways. Cancer Biol Ther 2005;4:139-163. https://doi.org/10.4161/cbt.4.2.1508 |
||||
| 153 Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L: Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997;7:821-830. https://doi.org/10.1016/S1074-7613(00)80400-8 |
||||
| 154 Schneider P, Bodmer JL, Holler N, Mattmann C, Scuderi P, Terskikh A, Peitsch MC, Tschopp J: Characterization of Fas (Apo-1, CD95)-Fas ligand interaction. J Biol Chem 1997;272:18827-18833. https://doi.org/10.1074/jbc.272.30.18827 |
||||
| 155 Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S: The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991;66:233-243. https://doi.org/10.1016/0092-8674(91)90614-5 |
||||
| 156 Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM: The receptor for the cytotoxic ligand TRAIL. Science 1997;276:111-113. https://doi.org/10.1126/science.276.5309.111 |
||||
| 157 Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, Holler N, Tschopp J: TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997;7:831-836. https://doi.org/10.1016/S1074-7613(00)80401-X |
||||
| 158 Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW: NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 1996;384:638-641. https://doi.org/10.1038/384638a0 |
||||
| 159 Guicciardi ME, Gores GJ: Life and death by death receptors. FASEB J 2009;23:1625-1637. https://doi.org/10.1096/fj.08-111005 |
||||
| 160 Goldar S, Khaniani MS, Derakhshan SM, Baradaran B: Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J Cancer Prev 2015;16:2129-2144. https://doi.org/10.7314/APJCP.2015.16.6.2129 |
||||
| 161 Zaman S, Wang R, Gandhi V: Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma 2014;55:1980-1992. https://doi.org/10.3109/10428194.2013.855307 |
||||
| 162 Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J: Inhibition of death receptor signals by cellular FLIP. Nature 1997;388:190-195. https://doi.org/10.1038/40657 |
||||
| 163 Billen LP, Shamas-Din A, Andrews DW: Bid: a Bax-like BH3 protein. Oncogene 2008;27:S93-104. https://doi.org/10.1038/onc.2009.47 |
||||
| 164 Pitti RM, Marsters SA, Lawrence DA, Roy M, Kischkel FC, Dowd P, Huang A, Donahue CJ, Sherwood SW, Baldwin DT, Godowski PJ, Wood WI, Gurney AL, Hillan KJ, Cohen RL, Goddard AD, Botstein D, Ashkenazi A: Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 1998;396:699-703. https://doi.org/10.1038/25387 |
||||
| 165 Bukau B, Weissman J, Horwich A: Molecular chaperones and protein quality control. Cell 2006;125:443-451. https://doi.org/10.1016/j.cell.2006.04.014 |
||||
| 166 Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E: HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J 1999;13:2061-2070. https://doi.org/10.1096/fasebj.13.14.2061 |
||||
| 167 Concannon CG, Orrenius S, Samali A: Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr 2001;9:195-201. https://doi.org/10.3727/000000001783992605 |
||||
| 168 Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G: Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 2001;3:839-843. https://doi.org/10.1038/ncb0901-839 |
||||
| 169 Gotoh T, Terada K, Oyadomari S, Mori M: hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ 2004;11:390-402. https://doi.org/10.1038/sj.cdd.4401369 |
||||
| 170 Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, Weichselbaum R, Nalin C, Alnemri ES, Kufe D, Kharbanda S: Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J 2000;19:4310-4322. https://doi.org/10.1093/emboj/19.16.4310 |
||||
| 171 Ghosh JC, Siegelin MD, Dohi T, Altieri DC: Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res 2010;70:8988-8993. https://doi.org/10.1158/0008-5472.CAN-10-2225 |
||||
| 172 Ghosh JC, Dohi T, Kang BH, Altieri DC: Hsp60 regulation of tumor cell apoptosis. J Biol Chem 2008;283:5188-5194. https://doi.org/10.1074/jbc.M705904200 |
||||
| 173 Lassot I, Robbins I, Kristiansen M, Rahmeh R, Jaudon F, Magiera MM, Mora S, Vanhille L, Lipkin A, Pettmann B, Ham J, Desagher S: Trim17, a novel E3 ubiquitin-ligase, initiates neuronal apoptosis. Cell Death Differ 2010;17:1928-1941. https://doi.org/10.1038/cdd.2010.73 |
||||
| 174 Magiera MM, Mora S, Mojsa B, Robbins I, Lassot I, Desagher S: Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ 2013;20:281-292. https://doi.org/10.1038/cdd.2012.124 |
||||
| 175 Wu X, Luo Q, Liu Z: Ubiquitination and deubiquitination of MCL1 in cancer: deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis 2020;11:556. https://doi.org/10.1038/s41419-020-02760-y |
||||
| 176 Huang NJ, Zhang L, Tang W, Chen C, Yang CS, Kornbluth S: The Trim39 ubiquitin ligase inhibits APC/CCdh1-mediated degradation of the Bax activator MOAP-1. J Cell Biol 2012;197:361-367. https://doi.org/10.1083/jcb.201111141 |
||||
| 177 Matsuura K, Huang NJ, Cocce K, Zhang L, Kornbluth S: Downregulation of the proapoptotic protein MOAP-1 by the UBR5 ubiquitin ligase and its role in ovarian cancer resistance to cisplatin. Oncogene 2017;36:1698-1706. https://doi.org/10.1038/onc.2016.336 |
||||
| 178 Chen C, Zhou Z, Ross JS, Zhou W, Dong JT: The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer 2007;121:80-87. https://doi.org/10.1002/ijc.22653 |
||||
| 179 Kim JB, Kim SY, Kim BM, Lee H, Kim I, Yun J, Jo Y, Oh T, Jo Y, Chae HD, Shin DY: Identification of a novel anti-apoptotic E3 ubiquitin ligase that ubiquitinates antagonists of inhibitor of apoptosis proteins SMAC, HtrA2, and ARTS. J Biol Chem 2013;288:12014-12021. https://doi.org/10.1074/jbc.M112.436113 |
||||
| 180 Ryu YS, Lee Y, Lee KW, Hwang CY, Maeng JS, Kim JH, Seo YS, You KH, Song B, Kwon KS: TRIM32 protein sensitizes cells to tumor necrosis factor (TNFalpha)-induced apoptosis via its RING domain-dependent E3 ligase activity against X-linked inhibitor of apoptosis (XIAP). J Biol Chem 2011;286:25729-25738. https://doi.org/10.1074/jbc.M111.241893 |
||||
| 181 Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M: The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006;124:601-613. https://doi.org/10.1016/j.cell.2006.01.021 |
||||
| 182 Gonzalvez F, Lawrence D, Yang B, Yee S, Pitti R, Marsters S, Pham VC, Stephan JP, Lill J, Ashkenazi A: TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell 2012;48:888-899. https://doi.org/10.1016/j.molcel.2012.09.031 |
||||
| 183 Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A: Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009;137:721-735. https://doi.org/10.1016/j.cell.2009.03.015 |
||||
| 184 Weber A, Heinlein M, Dengjel J, Alber C, Singh PK, Hacker G: The deubiquitinase Usp27x stabilizes the BH3-only protein Bim and enhances apoptosis. EMBO Rep 2016;17:724-738. https://doi.org/10.15252/embr.201541392 |
||||
| 185 Xu X, Liu J, Shen C, Ding L, Zhong F, Ouyang Y, Wang Y, He S: The role of ubiquitin-specific protease 14 (USP14) in cell adhesion-mediated drug resistance (CAM-DR) of multiple myeloma cells. Eur J Haematol 2017;98:4-12. https://doi.org/10.1111/ejh.12729 |
||||
| 186 Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM: Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010;463:103-107. https://doi.org/10.1038/nature08646 |
||||
| 187 Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, Wang X, Di W, Zhuang G: Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat Commun 2018;9:215. https://doi.org/10.1038/s41467-017-02693-9 |
||||
| 188 Zhou Z, Luo A, Shrivastava I, He M, Huang Y, Bahar I, Liu Z, Wan Y: Regulation of XIAP Turnover Reveals a Role for USP11 in Promotion of Tumorigenesis. EBioMedicine 2017;15:48-61. https://doi.org/10.1016/j.ebiom.2016.12.014 |
||||
| 189 Engel K, Rudelius M, Slawska J, Jacobs L, Ahangarian Abhari B, Altmann B, Kurutz J, Rathakrishnan A, Fernandez-Saiz V, Brunner A, Targosz BS, Loewecke F, Gloeckner CJ, Ueffing M, Fulda S, Pfreundschuh M, Trumper L, Klapper W, Keller U, Jost PJ, et al.: USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol Med 2016;8:851-862. https://doi.org/10.15252/emmm.201506047 |
||||
| 190 Xu M, Takanashi M, Oikawa K, Tanaka M, Nishi H, Isaka K, Kudo M, Kuroda M: USP15 plays an essential role for caspase-3 activation during Paclitaxel-induced apoptosis. Biochem Biophys Res Commun 2009;388:366-371. https://doi.org/10.1016/j.bbrc.2009.08.015 |
||||
| 191 Panner A, Crane CA, Weng C, Feletti A, Fang S, Parsa AT, Pieper RO: Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res 2010;70:5046-5053. https://doi.org/10.1158/0008-5472.CAN-09-3979 |
||||
| 192 Pickart CM: Mechanisms underlying ubiquitination. Annu Rev Biochem 2001;70:503-533. https://doi.org/10.1146/annurev.biochem.70.1.503 |
||||
| 193 Miyashita T, Harigai M, Hanada M, Reed JC: Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res 1994;54:3131-3135. | ||||
| 194 Sadot E, Geiger B, Oren M, Ben-Ze'ev A: Down-regulation of beta-catenin by activated p53. Mol Cell Biol 2001;21:6768-6781. https://doi.org/10.1128/MCB.21.20.6768-6781.2001 |
||||
| 195 Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS: APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol 2001;155:207-216. https://doi.org/10.1083/jcb.200105137 |
||||
| 196 Miyashita T, Reed JC: Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995;80:293-299. https://doi.org/10.1016/0092-8674(95)90412-3 |
||||
| 197 Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N: Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000;288:1053-1058. https://doi.org/10.1126/science.288.5468.1053 |
||||
| 198 Haupt Y, Maya R, Kazaz A, Oren M: Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296-299. https://doi.org/10.1038/387296a0 |
||||
| 199 Kubbutat MH, Jones SN, Vousden KH: Regulation of p53 stability by Mdm2. Nature 1997;387:299-303. https://doi.org/10.1038/387299a0 |
||||
| 200 Deveraux QL, Takahashi R, Salvesen GS, Reed JC: X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997;388:300-304. https://doi.org/10.1038/40901 |
||||
| 201 Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC: The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J 1997;16:6914-6925. https://doi.org/10.1093/emboj/16.23.6914 |
||||
| 202 Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD: Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000;288:874-877. https://doi.org/10.1126/science.288.5467.874 |
||||
| 203 Chen S, Fribley A, Wang CY: Potentiation of tumor necrosis factor-mediated apoptosis of oral squamous cell carcinoma cells by adenovirus-mediated gene transfer of NF-kappaB inhibitor. J Dent Res 2002;81:98-102. https://doi.org/10.1177/0810098 |
||||